



Périartérite noueuse
La périartérite noueuse (PAN) est une vascularite nécrosante systémique atteignant préférentiellement les vaisseaux de moyen calibre. Décrite en 1866 par Küssmaul et Maier, elle est parfois la conséquence d’une infection par le virus de l’hépatite B (PAN-VHB). Coïncidant avec les campagnes de vaccination contre le VHB, les nouveaux cas de PAN sont devenus très rares. Par contre, sont décrites d’autres formes secondaires de PAN, notamment au cours d’hémopathies et plus récemment une forme monogénique liée à des mutations de l’adénosine déaminase-2 (ADA2).
CLASSIFICATION et DIAGNOSTIC
Comme les critères de classification de l’American College of Rheumatology (ACR) ont été proposés avant le démembrement de la polyangéite microscopique, ils ne permettent pas de différencier la PAN et la polyangéite microscopique (Tableau 1) [1]. La nomenclature de Chapel Hill, basée sur la taille prédominante des vaisseaux atteints, a eu le mérite d’individualiser la polyangéite microscopique (Figure 1) [2]. Les vaisseaux de moyen calibre sont définis comme les principales artères viscérales et leurs branches de division initiale ainsi que leurs veines. (Figure 3). La PAN intéresse de façon prédominante les artères de moyen calibre et, parfois, des vaisseaux de calibre plus petit. L’atteinte rénale y est la conséquence d’une sténose ou occlusion des artères rénales de moyen calibre, se traduisant par des infarctus parenchymateux et/ou une hypertension artérielle rénovasculaire. La présence de nodules sous cutanés ou de microanévrysmes à l’artériographie oriente aussi vers une PAN. Les principaux éléments de diagnostic différentiel entre la PAN et la polyangéite microscopique sont résumés dans le Tableau 2. Les anticorps anti-cytoplasme des polynucléaires (ANCA) sont absents dans la PAN et leur présence permet d’exclure ce diagnostic. La présence d’une glomérulonéphrite, d’une hémorragie alvéolaire traduisant une capillarite pulmonaire ou d’ANCA oriente vers une polyangéite microscopique. Les critères de classification de l’ACR et la nomenclature de Chapel Hill ne doivent pas être utilisés à visée diagnostique. D’autres critères ont également été proposés [3].
ÉPIDÉMIOLOGIE
La PAN est une maladie devenue très rare. Elle touche autant d’hommes que de femmes, surtout entre 40 et 60 ans [4]. Les formes infantiles ne sont pas exceptionnelles. Son incidence a été estimée à 4 à 6 par million d’habitants en Grande- Bretagne [5]. Dans une population d’Esquimaux, largement infectés par le VHB, l’incidence était de 77 par million d’habitants [6]. En France, la prévalence de la maladie avait été estimée à 30,7 par million d’habitants adultes [7]. Les campagnes de vaccination contre le VHB et la diminution des pratiques à risque ont depuis entraîné une nette diminution de l’incidence de cette vascularite.
PATHOGÉNIE
Les lésions inflammatoires vasculaires endothéliales de la PAN-VHB résultent de dépôts de complexes immuns, au sein desquels la présence d’antigènes de surface du virus a été démontrée. La PAN pourrait ainsi être la conséquence d’une difficulté d’épuration par le système réticulo-endothélial de ces complexes immuns, qui pourraient déclencher l’activation du complément et ainsi la migration et l’activation des polynucléaires neutrophiles. Parmi les autres mécanismes envisagés, on évoque aussi une possible atteinte endothéliale directe par le virus, suivie d’une réplication in situ.
Il n’y a pas d’ANCA chez les patients atteints de PAN. Des anticorps anti-cellules endothéliales ont été mis en évidence mais leur rôle reste incertain. Un certain nombre de cytokines pourraient également intervenir dans la physiopathologie de la PAN. Une élévation de la concentration sérique d’interféron α (IFN-α), d’interleukine 2 (IL-2), et de façon plus discrète de TNF-α (tumor necrosis factor α) et d’IL-1β a été observée au cours de la PAN comme dans d’autres vascularites nécrosantes.
A part, les formes associées à des mutations perte de fonction d’ADA2, qui pourraient résulter d’une perte de l’intégrité de l’endothélium vasculaire et d’une polarisation des macrophages et monocytes vers un phénotype pro-inflammatoire [8,9].
ÉTIOLOGIE ET FACTEURS FAVORISANTS
La PAN classique est considérée comme idiopathique. Parmi les causes de PAN secondaires, la forme la plus classique est celle associée à une infection par le VHB. D’autres virus ou hémopathies peuvent être en cause. Une forme monogénique par mutations d’ADA2 peut aussi se présenter comme une PAN typique.
Périartérite noueuse due au virus de l'hépatite B
Autrefois, le mode de contamination le plus fréquent était par transfusion sanguine. Aujourd’hui, les rares contaminations sont d’origine sexuelle ou liées à une toxicomanie par voie intraveineuse. Dès 1970, un lien a été fait entre l’infection par le VHB et la maladie. Au début des années 1990, la moitié des cas de PAN était liée à une infection par le VHB. L’incidence de l’infection par le VHB et de la PAN-VHB a considérablement diminué, suite aux campagnes de vaccination et à une amélioration de la sécurité transfusionnelle. Dans les années 2000, les cas de PAN-VHB ne représentaient plus que 10 à 25 % des cas de PAN [10]. Ils sont actuellement devenus exceptionnels.
DADA2 (déficit en adénosine déaminase 2)
En 2014, deux études ont pu mettre en lien des cas familiaux de vasculopathie avec des mutations d’ADA2 [8,9]. Cette maladie monogénique est autosomique récessive, débute dans l’enfance ou chez l’adulte jeune, avec une grande variété de présentation clinique. Les patients avec DADA2 peuvent se présenter avec une maladie mimant une PAN comme des lésions histologiques de vascularite nécrosante, des anévrysmes des artères de moyen calibre, un livedo, des nodules, des atteintes neurologiques périphériques ou centrales et une hypo-gammaglobulinémie. Un début dans l’enfance, une forme familiale, une consanguinité, un accident vasculaire cérébral ischémique, une lymphopénie représentent des éléments d’orientation et incitent à rechercher la maladie. Le dosage d’ADA2 et la recherche de mutations permettent le diagnostic. La recherche systématique de mutations d’ADA2 dans les séries de patients initialement considérés comme ayant une PAN idiopathique a pu mettre en évidence entre 7,6 % à 31 % de patients atteints de DADA2 [11-13].
Autres causes
D’autres virus ont été associés à la survenue d’une PAN, mais ils ne rendent compte que de cas exceptionnels : virus de l’hépatite C, parvovirus B19, virus de l’immunodéficience humaine (VIH), du groupe herpès ou d’Epstein-Barr.
Outre ces agents infectieux, la PAN a parfois été décrite en association à des tumeurs malignes ou à des hémopathies, notamment la leucémie à tricholeucocytes. Les syndromes myélodysplasiques dont les leucémies myélomonocytaires chroniques et les anémies réfractaires peuvent aussi mimer une PAN classique.
ANATOMIE PATHOLOGIQUE
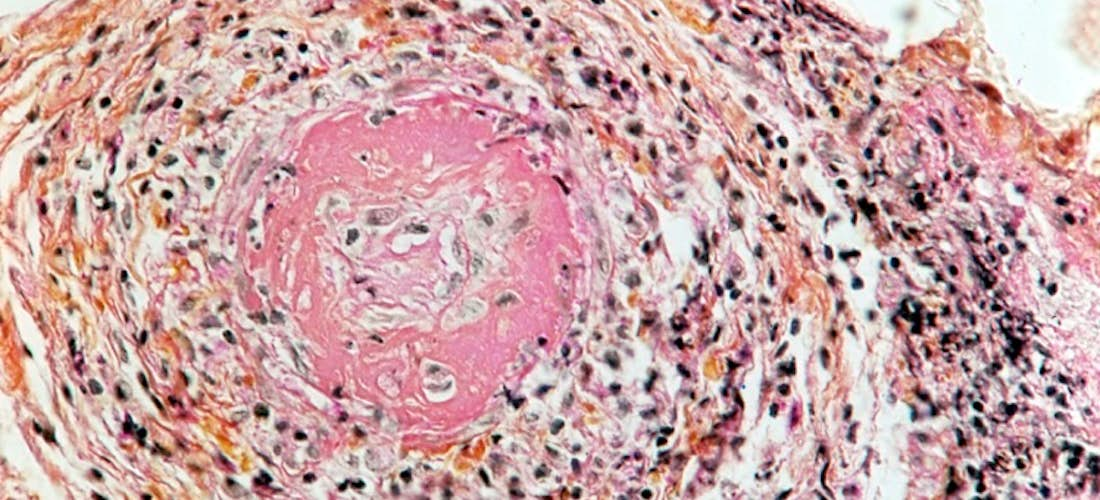
La lésion histologique élémentaire de la PAN est une vascularite nécrosante segmentaire, qui affecte préférentiellement les vaisseaux de moyen calibre avec une répartition ubiquitaire (Figure 5). Toutes les artères de l’organisme peuvent être touchées à l’exception de l’aorte et des artères pulmonaires. À la phase aiguë, l’inflammation de la paroi artérielle se caractérise par de la nécrose fibrinoïde de la media et une infiltration composée en majorité de polynucléaires neutrophiles à noyaux souvent pycnotiques, associés à un nombre variable de lymphocytes et d’éosinophiles. Les structures élastiques sont progressivement envahies par la nécrose fibrinoïde. L’atteinte de l’endothélium peut conduire à la thrombose et des microanévrysmes peuvent se développer. À un stade ultérieur, l’évolution se fait vers la cicatrisation avec une endartérite fibreuse qui rend impossible tout diagnostic histologique rétrospectif. Une autre donnée histologique caractéristique de la PAN est la coexistence de lésions de vascularite d’âges différents, avec des lésions fraîches côtoyant des lésions cicatricielles au sein du même tissu ou de tissus différents. Le caractère segmentaire impose l’épuisement des blocs des tissus prélevés à visée diagnostique, en l’absence de vascularite visualisée sur les premières coupes histologiques.
Il faut de préférence biopsier les sites touchés par la maladie comme la peau en cas de nodules ou l’artère temporale en cas de signes céphaliques. Chez les patients présentant des douleurs musculaires, la biopsie doit être pratiquée de préférence à un mollet ou à la loge antéro-externe de jambe. En cas de neuropathie périphérique, la biopsie neuromusculaire permet le diagnostic de vascularite nécrosante dans la majorité des cas. En cas d’intervention chirurgicale, notamment abdominale à la suite d’une perforation intestinale, d’une appendicite ou d’une cholécystite aiguë, l’analyse des pièces opératoires peut donner le diagnostic. La biopsie rénale est contre-indiquée car elle peut être responsable d’hématomes rénaux ou péri rénaux suite à la rupture de microanévrysmes.
MANIFESTATIONS CLINIQUES
Les principales manifestations cliniques de la PAN et leur fréquence sont résumées dans le Tableau 3 selon les principales séries de la littérature [4,14-18]. Les données des séries les plus anciennes sont à interpréter avec prudence car elles regroupaient souvent des cas de PAN et de polyangéite microscopique.
Signes généraux
Une altération de l’état général se rencontre dans plus deux tiers des cas. Un amaigrissement et une fièvre peuvent être les premiers signes et l’apparition des autres manifestations conduit alors au diagnostic.
Myalgies
Les myalgies sont présentes dans la moitié des cas. Elles sont intenses, diffuses, spontanées ou déclenchées par la pression. Parfois leur intensité peut en imposer pour une polymyosite mais les enzymes musculaires sont habituellement normales. Une amyotrophie est fréquente.
Arthralgies
Les arthralgies affectent la moitié des patients et peuvent être inaugurales. D’horaire inflammatoire, elles prédominent sur les grosses articulations. Les arthrites sont plus rares et non érosives.
Manifestations neurologiques
L’atteinte du système nerveux périphérique est fréquente et engage le pronostic fonctionnel. L’atteinte du système nerveux central est plus rare mais engage le pronostic vital.
Atteinte du système nerveux périphérique
L’atteinte du système nerveux périphérique est présente chez les trois quarts des patients et représente la manifestation inaugurale de la maladie dans 20 à 30 % des cas. Elle débute brutalement, parfois avec des œdèmes segmentaires. Des paresthésies et des douleurs peuvent précéder l’apparition d’une paralysie. Le tableau clinique le plus habituel est celui d’une mononeuropathie multiple sensitivomotrice avec un déficit évolutif dans le territoire du nerf péronier voire tibial. L’atteinte neurologique peut intéresser les membres supérieurs, dans les territoires ulnaire(s) ou radial(ux) voire médian(s). Une polyneuropathie distale, symétrique, sensitive ou sensitivomotrice est plus rare et s’observe dans 20 % des cas d’atteinte du système nerveux périphérique. L’atteinte des paires crâniennes est exceptionnelle et touche les nerfs III, VI, VII, VIII. Dans la plupart des cas, sous traitement, la mononeuropathie multiple s’améliore lentement mais le degré de récupération est très variable d’un malade à l’autre et demeure imprévisible. Les troubles sensitifs peuvent persister avec des paresthésies gênantes résiduelles.
L’électromyogramme est utile pour analyser le mécanisme et l’extension de la neuropathie. Il révèle le mécanisme axonal de la neuropathie en montrant des potentiels d’action moteurs et sensitifs absents ou diminués avec des vitesses de conduction nerveuse normales ou modérément allongées.
Atteinte du système nerveux central
Les localisations neurologiques centrales sont plus rares, de présentation variable mais d’origine ischémique.
Manifestations cutanées
Des manifestations cutanées sont rapportées chez la moitié des patients. Le purpura est possible, vasculaire et infiltré. Les nodules sous-cutanés témoignent de l’atteinte des vaisseaux de moyen calibre. Ils siègent sur les trajets vasculaires et prédominent aux membres inférieurs. Un livedo ramifié ou racemosa (reticularis pour les anglo-saxons) à larges mailles ouvertes est plus rare (Figure 8), de même que des ulcérations vasculaires ou une gangrène distale.
Atteinte rénale
La néphropathie vasculaire touche la moitié des patients. Son risque est l’évolution vers une insuffisance rénale. Une hypertension artérielle complique la néphropathie vasculaire chez le tiers des patients et est parfois sévère, voire maligne. Certains patients ont une néphropathie qui évolue favorablement après une période d’épuration extrarénale transitoire. Chez d’autres, une insuffisance rénale survient et conduit à la dialyse. Lorsque l’angiographie rénale est faite, elle peut montrer des sténoses segmentaires alternant avec des dilatations et/ou des microanévrysmes siégeant sur des branches des artères rénales (Figures 6).
Atteinte digestive
Les atteintes digestives sont l’une des manifestations les plus sévères et constituent la première cause de décès à 1 an au cours de la PAN. Au cours d’une étude reprenant 348 patients atteints de PAN, une atteinte digestive nécessitant le recours à la chirurgie au moment du diagnostic prédisait la mortalité de façon indépendante [4]. Les signes cliniques varient de la douleur abdominale isolée à des tableaux plus sévères, notamment chirurgicaux. Les hémorragies digestives et les perforations intestinales sont graves. Une atteinte de la vésicule biliaire (cholécystite) ou de l’appendice peut être la première manifestation clinique de PAN. La nécrose aiguë du pancréas ou le développement de kystes ou faux kystes pancréatiques sont retrouvés chez 2 à 3 % des patients [4]. L’atteinte pancréatique est souvent associée à une ischémie digestive et à des perforations qui peuvent être méconnues, expliquant son très mauvais pronostic. Le taux de survie à 5 ans des patients ayant ces manifestations digestives n’est que de 50 % [7].
Atteinte cardiaque
L’atteinte cardiaque touche un patient sur cinq. La PAN lèse essentiellement le myocarde soit par le biais d’une vascularite des artères coronaires ou de leurs branches, soit par celui d’une hypertension artérielle sévère non contrôlée. L’atteinte péricardique est rare. Une insuffisance cardiaque peut survenir. Une cardiomyopathie est retrouvée dans 7,5 % des cas [4]. Une tachycardie est fréquente et des troubles du rythme, principalement supraventriculaire, ou de la conduction sont parfois observés. L’occlusion d’une artère distale des membres peut entraîner une gangrène des orteils ou des doigts. L’angiographie montre la présence de sténoses ou de microanévrysmes. Le phénomène de Raynaud peut être isolé ou compliqué de nécrose.
Autres atteintes
Les poumons sont habituellement épargnés au cours de la PAN et une atteinte pulmonaire fait habituellement remettre en cause le diagnostic ou rechercher une infection surajoutée.
Une orchite non infectieuse est une des manifestations caractéristiques de la PAN chez l’homme au point d’avoir été incluse dans les critères de classification de l’ACR [1]. Elle est plus fréquente chez les patients atteints de PAN-VHB.
FORMES CLINIQUES DE PAN
Formes localisées de PAN
La PAN peut rester strictement localisée à la peau, s’accompagnant éventuellement de quelques manifestations neurologiques périphériques et/ou articulaires peu marquées. Ces formes sont bénignes mais rechutent volontiers.
Manifestations spécifiques de la PAN-VHB
La PAN-VHB survient habituellement dans les semaines ou mois qui suivent la contamination. L’hépatite est habituellement silencieuse ou ne se traduit que par une augmentation modérée des transaminases. Les manifestations cliniques de la PAN-VHB ne sont pas différentes de celles de la PAN non liée à l’infection par le VHB (Tableau 4). Toutefois, les patients présentant une PAN-HB se présentent avec une maladie plus sévère : plus souvent d’atteinte neurologique périphérique, d’atteinte digestive, de cardiomyopathie spécifique, d’hypertension artérielle sévère et d’orchite [4]. Ils rechutent moins souvent.
Périartérite noueuse pédiatrique
Une classification européenne des vascularites de l’enfant a été proposée [19]. Les formes pédiatriques de PAN doivent faire rechercher un DADA2. En dehors de cette maladie monogénique, de très rares formes de PAN sont décrites dans l’enfance. L’âge moyen au diagnostic est de 10 ans avec un sex-ratio en faveur des garçons [20]. Les manifestations cliniques de la PAN chez l’enfant sont comparables à celles de l’adulte, à l’exception des formes cutanées qui paraissent plus fréquentes mais avec une évolution comparable. Dans les formes systémiques, la PAN de l’enfant a la même gravité que celle de l’adulte mais avec des rechutes plus fréquentes, souvent mineures et cutanées, qui motivent volontiers la poursuite d’un traitement.
INVESTIGATIONS COMPLÉMENTAIRES
Le diagnostic de PAN repose habituellement sur une preuve histologique. Une biopsie musculaire et/ou neuromusculaire permet un diagnostic histologique dans les trois quarts des cas. La biopsie rénale est déconseillée. Les biopsies cutanées montrent une vascularite dans la moitié des cas, le plus souvent leucocytoclasique et souvent nécrosante. Quand une biopsie ne peut être réalisée, le diagnostic peut être confirmé par l’angiographie.
Examens biologiques
Aucun examen biologique n’est spécifique de la PAN. Un syndrome inflammatoire est présent chez les trois quarts des patients. Une hyperéosinophilie modérée peut être retrouvée, exceptionnellement supérieure à 1500/mm3. Une infection par le VHB doit être systématiquement recherchée par un test sérologique, puis une quantification de son ADN (acide désoxyribonucléique) en cas de positivité. La présence d’ANCA constitue un critère d’exclusion du diagnostic.
Angiographies
Bien que non pathognomonique, la présence de microanévrysmes et de sténoses irrégulières des artères de moyen calibre à l’artériographie rénale et coelio-mésentérique est très évocatrice de PAN (Figures 6). La fréquence de ces anomalies, focales, segmentaires et fluctuantes dans le temps, est de 90 % en cas de signes digestifs et de 40 à 60 % en leur absence. Ces anomalies disparaissent avec le traitement de la vascularite mais un infarctus viscéral peut être la conséquence directe de l’obstruction artérielle.
ÉVOLUTION
La PAN est habituellement une maladie aiguë qui, dans sa forme systémique, se manifeste typiquement par une poussée unique, mais parfois sévère, voire mortelle si le traitement adéquat n’est pas rapidement prescrit.
Depuis l’utilisation des corticoïdes puis leur association aux immunosuppresseurs dans les formes sévères, et les traitements antiviraux combinés aux échanges plasmatiques dans les formes liées au VHB, le pronostic s’est amélioré, avec un taux de survie à 5 et 10 ans de 83 et 74 % respectivement pour la PAN et de 73 et 60 % respectivement pour la PAN-VHB [4]. Le taux de survie sans rechute à 5 ans est, respectivement, de 60 % et 67 % pour les PAN et les PAN-VHB [4].
Rechutes
Une fois la rémission obtenue, les rechutes sont classiquement peu fréquentes au cours de la PAN. Dans une étude portant sur 348 patients atteints de PAN suivis pendant 68 mois en moyenne, 10,6 % des PAN-VHB et 28 % des PAN ont rechuté, dans un délai moyen de respectivement 43 et 26 mois par rapport à la première poussée [4].
Décès
Décès attribuables à la vascularite
Un certain nombre de patients meurent durant les premiers mois de la maladie d’atteinte multiviscérale non contrôlable par les traitements. La plupart de ces décès sont la conséquence d’une atteinte digestive.
Décès imputables aux traitements
Les traitements peuvent aussi être responsables d’un certain nombre d’effets secondaires, certains sévères, éventuellement responsable de décès. Les infections bactériennes avec septicémies représentent la première cause de mortalité liée aux traitements. Les infections virales surviennent plus tardivement, favorisées par l’immunodépression profonde induite par les immunosuppresseurs. De rares cas de pneumonie à Pneumocystis jiroveci ont été rapportés, moins fréquemment qu’au cours de la granulomatose avec polyangéite.
TRAITEMENT
PRINCIPES DU TRAITEMENT
Le traitement initial est adapté aux critères de sévérité du FFS [21-23]. Le FFS tient compte des manifestations associées à une surmortalité, à savoir l’existence d’un âge supérieur à 65 ans, d'une cardiomyopathie spécifique, de manifestations gastro-intestinales et d’une insuffisance rénale définie par une créatininémie stabilisée supérieure à 150 µmol /L (Tableau 5) [23].
Les corticoïdes
Le traitement initial de la PAN, sans infection par le VHB, comprend une corticothérapie. La dose initiale est de 1 mg/kg/j sans dépasser 60 mg/j. Elle peut parfois être précédée, selon la gravité, d’un à trois bolus de méthylprednisolone (15 mg/kg/j). Après un traitement initial de 3 semaines, les corticoïdes doivent être diminués rapidement pour atteindre approximativement 20 mg/j à 3 mois, 10 mg/j à 6 mois et 5 mg/j à 1 an.
Le cyclophosphamide
La prescription d’immunosuppresseurs au cours de la PAN sans infection par le VHB, et notamment de cyclophosphamide, est réservée aux formes sévères, c’est-à-dire avec existence d’au moins un des facteurs de mauvais pronostic du FFS [23]. Le cyclophosphamide est administré par trois bolus de 0,6 g/m2 espacés de 14 jours puis trois bolus supplémentaires espacés de 21 jours, à la posologie de 0,7 g/m2. Il est recommandé de ne pas dépasser 1200 mg par bolus et de diminuer la posologie de cyclophosphamide chez les personnes âgées de plus de 65 ans et en cas d’insuffisance rénale [21,22]. Une hydratation abondante et l’utilisation de mesna doivent être associées au bolus.
Autres traitements immunosuppresseurs
D’autres immunosuppresseurs peuvent être utilisés mais n’ont pas été validés. Ils peuvent constituer une alternative au cyclophosphamide dans certaines situations, notamment en cas de rechute. Ils ont surtout un intérêt en relais du cyclophosphamide, une fois la rémission obtenue, au cours de la PAN non liée au VHB. L’azathioprine est le traitement d’entretien recommandé de première intention, dès que la rémission a été obtenue. La dose habituelle est de 2 à 3 mg/kg/j. Le méthotrexate peut aussi être prescrit une fois la rémission obtenue. La durée recommandée du traitement est de 12 à 18 mois.
Echanges plasmatiques
Les échanges plasmatiques n’améliorent pas le taux de survie au cours de la PAN non liée à une infection virale. En revanche, ils sont utiles dans le traitement de la PAN-VHB avec des bénéfices démontrés [24].
Traitements adjuvants
Une prophylaxie par le cotrimoxazole est systématique, pour prévenir la pneumonie à Pneumocystis jiroveci, pendant la durée du traitement par cyclophosphamide et tant que le taux de lymphocytes T CD4+ est inférieur à 250/mm3 (par exemple, Bactrim© forte 800 mg : 1 comprimé, 3 fois par semaine ou Bactrim© 400 mg : 1 comprimé/jour). L'ostéoporose cortisonique doit être combattue, selon les recommandations, par une activité physique régulière, une alimentation suffisamment riche en calcium, une supplémentation vitamino-calcique et un bisphosphonate, si la masse osseuse est initialement basse, débutés dès que la corticothérapie est instituée. La kinésithérapie est également nécessaire chez les malades présentant une neuropathie périphérique. La pression artérielle doit être contrôlée et les inhibiteurs de l’enzyme de conversion de l’angiotensine ou des récepteurs de l’angiotensine II sont prescrits dans l’hypertension artérielle rénovasculaire. La correction d’un état nutritionnel médiocre peut aider à diminuer l’incidence des infections induites par les traitements cytotoxiques. Chez les patients âgés, la minimisation de la dose de corticoïdes et d’immunosuppresseurs permet d’améliorer le pronostic vital, essentiellement en réduisant le nombre d’effets secondaires des médicaments [25].
STRATÉGIES THÉRAPEUTIQUES
Le traitement de la PAN diffère selon que la maladie est primitive ou secondaire à une infection par le VHB, à un autre agent infectieux voire à une maladie monogénique.
Traitement de la périartérite noueuse primitive
Le traitement recommandé chez les patients dont le FFS est égal à 0 repose sur la corticothérapie isolée en première intention [21-23]. Les immunosuppresseurs ne sont associés qu’en cas d’échec de ce traitement ou de rechute, situation qui concerne 40 % des patients, après 5 ans de suivi. Un traitement associant d’emblée corticoïdes et cyclophosphamide permettrait de réduire ce taux entre 15 et 20 %, mais n’est pas recommandé car à l’origine d’effets secondaires chez 40 à 60 % d’entre eux. Dans les formes sévères (FFS ≥ 1), l’association de corticoïdes et de cyclophosphamide est indispensable d’emblée et améliore le pronostic vital [26-28].
Traitement de la PAN-VHB
Dans la PAN-VHB, le traitement conventionnel par corticothérapie et cyclophosphamide est délétère. Il favorise la persistance du virus et sa réplication et donc l’évolution vers une hépatite chronique et la cirrhose. La stratégie thérapeutique est la suivante [24] : corticothérapie initiale brève permettant de contrôler les manifestations les plus sévères de la PAN, puis arrêt brutal des corticoïdes facilitant la séroconversion ; adjonction d’échanges plasmatiques afin de mieux contrôler le cours évolutif de la PAN ; combinaison aux antiviraux (vidarabine autrefois, puis plus récemment interféron α, lamivudine, adéfovir, entécavir, nouveaux antiviraux). Sous traitement, le pronostic vital est favorable et le taux de séroconversion HBe/ antiHBe augmente notablement (> 50 % des cas).
Traitement des DADA2
Le traitement des formes monogéniques symptomatiques de DADA2 repose sur les anti-TNF qui doivent être instaurés dès le diagnostic. Ils préviennent les complications neurologiques ischémiques centrales qui en font la gravité [26,29].
A RETENIR
Quand évoquer le diagnostic de périartérite noueuse (PAN) ?
Le diagnostic doit être évoqué devant les signes suivants, d’autant plus qu’ils sont associés :
- Altération de l’état général
- Neuropathie périphérique
- Arthralgies d’allure inflammatoire
- Myalgies
- Purpura vasculaire ou nodules
- Atteinte rénale vasculaire
Xavier PUÉCHAL
Bibliographie
- Lightfoot RW, JR, Michel BA, Bloch DA et al. The American College of Rheumatology 1990 criteria for the classification of polyarteritis nodosa. Arthritis Rheum 1990, 33 : 1088-93.
- Jennette JC, Falk RJ, Bacon PA et al. 2012 revised International Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum 2013, 65 : 1-11.
- Henegar C, Pagnoux C, Puéchal X et al. A paradigm of diagnostic criteria for polyarteritis nodosa : analysis of a series of 949 patients with vasculitides. Arthritis Rheum 2008, 58 : 1528-38.
- Pagnoux C, Seror R, Henegar C et al. Clinical features and outcomes in 348 patients with polyarteritis nodosa : a systematic retrospective study of patients diagnosed between 1963 and 2005 and entered into the French Vasculitis Study Group database. Arthritis Rheum 2010, 62 : 616-26.
- Scott DG, Bacon PA, Elliott PJ et al. Systemic vasculitis in a district general hospital 1972-1980 : clinical and laboratory features, classification and prognosis of 80 cases. Q J Med 1982, 51 : 292-311.
- McMahon BJ, Heyward WL, Templin DW et al. Hepatitis B-associated polyarteritis nodosa in Alaskan Eskimos : clinical and epidemiologic features and long-term follow-up. Hepatology 1989, 9 : 97-101.
- Mahr A, Guillevin L, Poissonnet M, Ayme S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener’s granulomatosis, and Churg- Strauss syndrome in a French urban multiethnic population in 2000 : a capture- recapture estimate. Arthritis Rheum 2004, 51 : 92-9.
- Zhou Q, Yang D, Ombrello AK, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med 2014 ; 370 : 911-20.
- Navon Elkan P, Pierce S B, Segel R, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med 2014 ; 370 : 921-31.
- Guillevin L, Mahr A, Callard P et al. Hepatitis B virus-associated polyarteritis nodosa : clinical characteristics, outcome, and impact of treatment in 115 patients. Medicine (Baltimore) 2005, 84 : 313-22.
- Schnappauf O, Sampaio Moura N, et al. Sequence-based screening of patients with idiopathic polyarteritis nodosa, granulomatosis with polyangiitis, and microscopic polyangiitis for deleterious genetic variants in ADA2. Arthritis Rheumatol 2021 ; 73 : 512-9.
- Schepp J, Proietti M, Frede N, et al. Screening of 181 patients with antibody deficiency for deficiency of adenosine deaminase 2 sheds new light on the disease in adulthood. Arthritis Rheumatol 2017 ; 69 : 1689-1700.
- Caorsi R, Penco F, Grossi A, et al. ADA2 deficiency (DADA2) as an unrecognised cause of early onset polyarteritis nodosa and stroke: a multicentre national study. Ann Rheum Dis 2017 ;76 : 1648-56.
- Frohnert PP, Sheps SG. Long-term follow-up study of periarteritis nodosa. Am J Med 1967, 43 : 8-14.
- Leib ES, Restivo C, Paulus HE. Immunosuppressive and corticosteroid therapy of polyarteritis nodosa. Am J Med 1979, 67 : 941-7.
- Cohen RD, Conn DL, Ilstrup DM. Clinical features, prognosis, and response to treatment in polyarteritis. Mayo Clin Proc 1980, 55 : 146-55.
- Guillevin L, Le Thi Huong D, Godeau P et al. Clinical findings and prognosis of polyarteritis nodosa and Churg-Strauss angiitis : a study in 165 patients. Br J Rheumatol 1988, 27 : 258-64.
- Fortin PR, Larson MG, Watters AK et al. Prognostic factors in systemic necrotizing vasculitis of the polyarteritis nodosa group : a review of 45 cases. J Rheumatol 1995, 22 : 78-84.
- Ozen S, Pistorio A, Iusan SM, et al. EULAR/PRINTO/PRES criteria for Henoch-Schonlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann Rheum Dis 2010 ;69 : 798-806.
- Iudici M, Quartier P, Pagnoux C, et al. Childhood- versus adult-onset polyarteritis nodosa. Results from the French Vasculitis Study Group registry. Autoimmun Rev 2018 ; 17 : 984-9.
- Haute Autorité de Santé. Protocole National de Diagnostic et de Soins. Vascularites nécrosantes systémiques (périartérite noueuse et vascularites associées aux ANCA), 2019. https://www.has-sante.fr/upload/docs/application/pdf/2019-06/pnds_vns.pdf
- Terrier B, Darbon R, Durel CA, et al. French recommendations for the management of systemic necrotizing vasculitides (polyarteritis nodosa and ANCA-associated vasculitides). Orphanet J Rare Dis 2020 ; 15(Suppl 2) : 351.
- Guillevin L, Pagnoux C, Seror R et al. The five-factor score revisited : assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore) 2011 ; 90 : 19-27.
- Guillevin L, Lhote F, Cohen P et al. Polyarteritis nodosa related to hepatitis B virus. A prospective study with long-term observation of 41 patients. Medicine (Baltimore) 1995 ; 74 : 238-53.
- Pagnoux C, Quéméneur T, Ninet J, et al. Treatment of systemic necrotizing vasculitides in patients aged sixty-five years or older: results of a multicenter, open-label, randomized controlled trial of corticosteroid and cyclophosphamide-based induction therapy. Arthritis Rheumatol 2015 ; 67 : 1117-27.
- Chung SA, Gorelik M, Langford CA, et al. 2021 American College of Rheumatology/Vasculitis Foundation guideline for the management of polyarteritis nodosa. Arthritis Rheumatol 2021 Jul 8. doi: 10.1002/art.41776 : sous presse.
- Guillevin L, Cohen P, Mahr A, et al. Treatment of polyarteritis nodosa and microscopic polyangiitis with poor prognosis factors: a prospective trial comparing glucocorticoids and six or twelve cyclophosphamide pulses in sixty-five patients. Arthritis Rheum 2003 ; 49 : 93-100.
- Samson M, Puéchal X, Mouthon L, et al. Microscopic polyangiitis and non-HBV polyarteritis nodosa with poor-prognosis factors: 10-year results of the prospective CHUSPAN trial. Clin Exp Rheumatol 2017 ; 35 Suppl 103 : 176-84.
- Ombrello AK, Qin J, Hoffmann PM, et al. Treatment strategies for deficiency of adenosine deaminase 2. N Engl J Med 2019 ; 380 : 1582-4.22. Guillevin L, Pagnoux C, Seror R et al. The five-factor score revisited : assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore) 2011, 90 : 19-27.
Figure 1. Nomenclature révisée de la Conférence de Chapel Hill

Figure 2. Chapel Hill Vascularites des gros vaisseaux
Figure 3. Chapel Hill Vascularites des vaisseaux de moyen calibre
Figures 4. Chapel Hill Vascularite des petits vaisseaux
Figures 5. HISTOLOGIE VASCULARITES
Figures 6. RADIOLOGIE PAN
Tableau 1. Critères de classification de la PAN
Tableau 2. Diagnostic différentiel entre PAN et PAM
Tableau 3. Manifestations cliniques de la PAN
Tableau 4. Manifestations cliniques de la PAN HBV
Tableau 5. FFS Facteurs pronostiques des vascularites nécrosantes systémiques
Figure 8. Livedo racemosa




