



Granulomatose éosinophilique avec polyangéite
Un cours enregistré est également disponible en cliquant ici pour la présentation générale, et ici sur les immunothérapies ciblées au cours de la GEPA.
La granulomatose éosinophilique avec polyangéite (GEPA) (Churg-Strauss) a été individualisé en 1951 à partir d’une série autopsique de patients ayant présenté un tableau anatomo-clinique voisin de celui de la périartérite noueuse (PAN) (1). Les caractéristiques distinctives sont la présence d’un asthme, d’une éosinophilie et les constatations anatomo-pathologiques qui comprennent une vascularite des vaisseaux de petit calibre, artérioles et veinules, et un granulome (1).
CLASSIFICATION
Différents systèmes de classification ont été proposés. Les critères de classification de la GEPA de l’American College of Rheumatology (ACR) sont indiqués dans le tableau 1 (2). Selon la nomenclature de Chapel Hill, la GEPA fait partie des vascularites touchant les vaisseaux de petit calibre (Figure 1 et Figures 4) (3). Lanham a décrit d’autres critères qui sont souvent utilisés à visée diagnostique (4). Ils comprennent l’association d’un asthme, d’une éosinophilie supérieure à 1.500/mm3 et de symptômes de vascularite systémique atteignant au moins deux sites extra-pulmonaires. Comme un certain nombre de vascularites qui touchent les petits vaisseaux, la GEPA est assez souvent associée à la présence d’anticorps anti-cytoplasme des polynucléaires neutrophiles (ANCA). Mais d’une part, la prévalence des ANCA est bien moindre que dans les autres vascularites associées aux ANCA : les ANCA ne sont présents que dans 30 à 40 % des cas de GEPA. D’autre part, contrairement à la granulomatose avec polyangéite où les ANCA ont typiquement une fluorescence cytoplasmique (cANCA) et sont dirigés contre la protéinase 3 (PR3), les ANCA observés au cours de la GEPA ont habituellement une fluorescence périnucléaire (pANCA) et pour cible la myéloperoxidase (MPO) (Figure 7), comme au cours de la polyangéite microscopique (PAM).
MECANISMES
La pathogénie de la GEPA reste mal connue (5-7). On évoque la responsabilité éventuelle d’antigènes inhalés ou de certaines stimulations antigéniques comme facteur déclenchant (cf 8 - Facteurs déclenchants). La GEPA est classiquement une affection Th2 avec une activation et une déviation de la balance des lymphocytes T (Figure 9). Les lymphocytes T de patients avec GEPA peuvent produire de grande quantité de cytokines Th2 comme l’interleukine (IL)-4 et l’IL-13. L’IL-5 est aussi fortement activée lors des phases d’activité de la GEPA et est particulièrement utile pour l’activation, la maturation et la survie des éosinophiles. Cependant, le phénotype clinique ne peut être uniquement expliqué par une réponse Th2 exagérée. Les lymphocytes Th1 et Th17 sont aussi impliquées et secrètent de forte quantité d’IL-17A dans les phases tardives de la maladie. De plus, les arguments s’accumulent pour attribuer un rôle aux lymphocytes B et à la réponse humorale comme facteur contribuant à la pathogénie de la GEPA. Des taux élevés de cellules B activées et de B mémoires sont trouvés en phase active. Des anticorps de fluorescence périnucléaire et de spécificité anti-MPO sont présents chez 30 à 40 % des patients avant traitement. Les patients avec des poussées de GEPA ont souvent des taux élevés d’IgE sériques et des complexes immuns contenant des IgE, confortant l’hypothèse pathogénique d’une vascularite à complexes immuns induite par un allergène. Des taux élevés d’IgG4 sont aussi fréquemment retrouvés et la commutation vers la production d’IgG4 est liée au milieu inflammatoire ambiant conditionnant la maturation des cellules B, et particulièrement à la présence de cytokines Th2 comme l’IL4, IL5 and IL13. Les éosinophiles activés pourraient à leur tour activer les cellules endothéliales, engendrer les lésions inflammatoires vasculaires et constituer l’infiltrat extravasculaire. Par ailleurs, le relargage des protéines cationiques de l’éosinophile pourrait participer à la cardiotoxicité. Il n'existe aucun moyen de prévoir au sein des patients asthmatiques ceux qui seraient susceptibles de développer une GEPA (8).
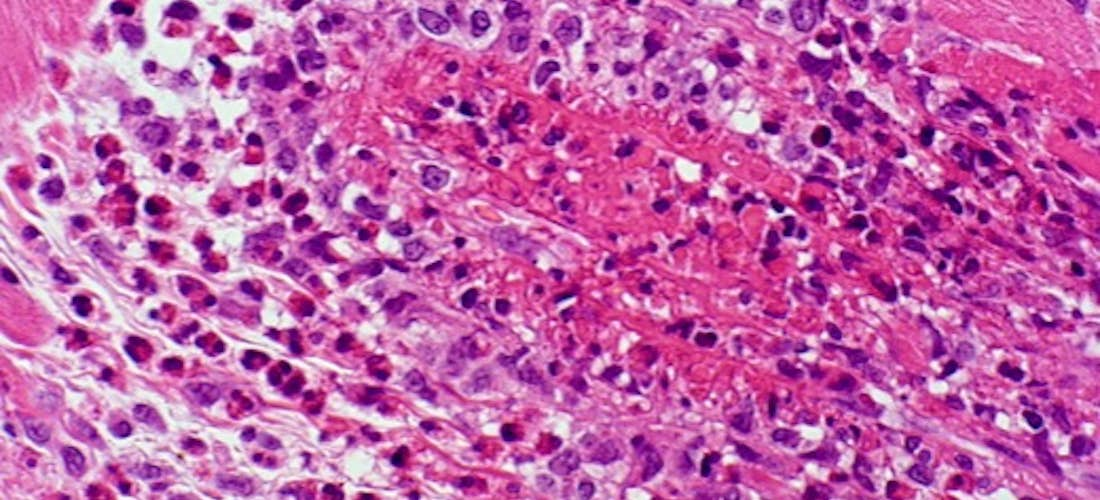
ANATOMIE PATHOLOGIQUE
Il existe 3 types de lésions élémentaires dans la GEPA dont la coexistence est rare :
- La vascularite nécrosante touche les artères et les veines de petit calibre avec des lésions segmentaires et un infiltrat riche en éosinophiles
- Une infiltration éosinophilique tissulaire
- Des granulomes extravasculaires contenant une nécrose centrale entourée de cellules épithélioïdes.
EPIDEMIOLOGIE
La GEPA est une maladie rare et peu de grandes séries ont été publiées (9-11). Selon les données anatomo-pathologiques de Lie, sur 1337 patients autopsiés atteints de vascularite, 40 seulement présentaient une GEPA (12). Au Royaume-Uni, dans le Comté de Norwich, l’incidence annuelle de GEPA a été estimée à 2,4/million d’habitants entre 1988 et 1994 avec une prédominance rurale (13). En effet, l’incidence rurale était de 5,1/million d’habitants versus 1,8 pour les zones urbaines. Cette prédominance a fait suspecter la responsabilité de pollens ou de pesticides. Néanmoins, les travaux les plus récents de la même équipe semblent nuancer la prédominance rurale de la GEPA (14). L’incidence de la GEPA en Espagne et en Norvège a été estimée respectivement à 0,9 (0,1-3,2) et 0,5 (0,06-1,8) par million d’habitants (15). Dans les séries de vascularites dénommées autrefois du “groupe de la périartérite noueuse” (16), 10 à 26 % des patients présentaient une GEPA (17-21). Dans les études prospectives menées par le Groupe Français d'Etude des Vascularites, la GEPA représente 19 % des cas (19,22,23). Une étude épidémiologique française menée dans le département de la Seine-Saint-Denis a permis d’objectiver une prévalence de 7,3 (2,8-14,4) et une incidence annuelle de 0,9 (0,03-5,6) par million d’habitants de plus de 15 ans (24).
La GEPA peut toucher des sujets de tous âges, avec une incidence maximale entre 30 et 50 ans. Il existe d’exceptionnelles formes pédiatriques. L’âge moyen lors du diagnostic était de 50 ans dans notre série de 338 patients (11), ce qui est plus bas que celui observé au cours de la PAN ou surtout de la PAM. Le sexe/ratio est proche de 1 (9,11).
MANIFESTATIONS CLINIQUES
La GEPA évolue habituellement en 3 phases :
- Initialement le patient présente un asthme et différentes manifestations allergiques
- Dans un second temps, une éosinophilie et un infiltrat pulmonaire s’installent
- Puis, le plus souvent plusieurs années après l’apparition de l’asthme, surviennent les différentes manifestations systémiques de la GEPA.
Les enquêtes systématiques font apparaître des antécédents personnels ou familiaux d’allergie dans respectivement 64 et 25 % des cas (9,25). Il est souvent difficile de dater précisément l’entrée dans la maladie. Les manifestations cliniques apparaissent en quelques semaines avec une aggravation progressive de l’asthme accompagnée de signes généraux : fièvre, amaigrissement, asthénie. Puis au bout de quelques semaines, les symptômes systémiques surviennent. Les principales manifestations cliniques de la GEPA et leur fréquence sont synthétisées dans le tableau 2 (4,9,25-30).
Manifestations pulmonaires
Asthme
L’asthme est présent dans 91 % des cas et est au premier plan du tableau clinique. Il est un des éléments essentiels pour évoquer le diagnostic (11). Cependant, de façon exceptionnelle, des patients peuvent se présenter avec des équivalents allergiques (rhinites, allergies cutanées, ...) ou voir apparaître l’asthme de façon concomitante avec les autres manifestations systémiques de la vascularite. L’asthme apparaît en moyenne 9 +/- 10 ans avant les premiers signes de vascularite (9,11). Souvent, il s’agit d’un asthme à début tardif, survenant autour de la quarantaine, même si l’asthme peut avoir connu un début infantile. Chez la moitié des patients, l’asthme est sévère et nécessite une corticothérapie locale inhalée ou administrée par voie générale. L’asthme s’aggrave dans les semaines précédant l’apparition de la vascularite, devient cortico-dépendant et nécessite souvent une hospitalisation pour traiter une attaque ou une insuffisance respiratoire. Bien que sévère, l’asthme n’est que rarement le motif du décès. Lorsque la vascularite survient, l’asthme est habituellement contrôlé par la corticothérapie et passe au second plan derrière les autres manifestations de vascularite.
Infiltrat pulmonaire
La radiographie pulmonaire est anormale dans 38 à 70 % des cas (4,11). Les opacités sont uni- ou bilatérales mais surtout transitoires, labiles (Figure 10). Quand elles se localisent à la périphérie du parenchyme pulmonaire, le diagnostic différentiel est celui d’une pneumonie chronique à éosinophiles. Les opacités régressent spontanément ou sous corticothérapie. Ils sont habituellement composés d’éosinophiles mais peuvent beaucoup plus rarement traduire une hémorragie alvéolaire.
Pleurésie
Dans les études rétrospectives, une pleurésie était notée dans 9 à 65 % des cas (4,11) mais une fréquence bien moindre de l’ordre de 3 % a été trouvée dans une méta-analyse d’études prospectives. L’atteinte pleurale peut être uni- ou bilatérale et est souvent asymptomatique. L’analyse du liquide révèle un exsudat. La principale caractéristique du liquide pleural est la richesse en polynucléaires éosinophiles qui peuvent représenter plus de 85 % des globules blancs. A l’opposé, un épanchement transudatif peut témoigner d’une insuffisance cardiaque.
Une vascularite et une infiltration de la plèvre par des éosinophiles peuvent être observées. Le diagnostic de GEPA est rarement évoqué sur les constatations d’une vascularite à la biopsie pleurale (9).
Hémorragie alvéolaire
Comme au cours des autres vascularites touchant les petits vaisseaux, une capillarite pulmonaire peut survenir et se caractérise par des hémorragies alvéolaires chez 4 % des patients (11,31,32). Le diagnostic est évoqué devant l’association d’hémoptysies, d’un infiltrat pulmonaire et d’une anémie (Clichés 6 et 7). La tomodensitométrie pulmonaire peut aider au diagnostic dans les cas difficiles, de même que le lavage alvéolaire. Le diagnostic est confirmé au lavage alvéolaire lorsqu’il revient uniformément rouge ou rosé et/ou si le lavage revient clair lorsque la coloration de Perls met en évidence plus de 30 % de sidérophages (macrophages positifs) et/ou le score de Golde est supérieur à 100.
Autre atteinte pulmonaire
Une paralysie phrénique a été exceptionnellement décrite et pourrait être en rapport avec une atteinte ischémique vasculaire du nerf phrénique (33).
Manifestations neurologiques
Atteinte du système nerveux périphérique
Une neuropathie périphérique affecte 46 à 75 % des patients atteints de GEPA (4,9-11,18,19,25,27,34). Les douleurs à type de brûlure ou de paresthésie peuvent précéder les premiers signes objectifs déficitaires sensitifs et/ou moteurs. Une mononeuropathie multiple atteint préférentiellement les membres inférieurs et surtout le nerf nerf fibulaire commun voire tibial et plus rarement le radial, cubital et/ou médian avec une atteinte sensitive et/ou motrice. Une atteinte du nerf fibulaire commun était présente chez 66 % de nos patients et chez 83 % de ceux qui présentaient une mononeuropathie multiple (9). Les paralysies sont rapidement progressives au début de la maladie. Seule la sensibilité superficielle est atteinte dans les formes discrètes. Une polyneuropathie sensitive ou sensitivomotrice est possible. Les nerfs crâniens sont moins souvent affectés mais une atteinte du II, III, VI et VIII peut être observée. Dans de rares cas, l’atteinte neurologique périphérique est bilatérale et symétrique et peut en imposer pour un syndrome de Guillain-Barré (35). Une neuropathie périphérique n’est pas associée à une surmortalité. Au cours des premiers mois de la maladie, les atteintes du SNP peuvent se succéder dans le temps et dans l’espace. Après un certain temps, les paralysies régressent mais le degré de récupération est imprévisible. Des séquelles motrices majeures peuvent s’observer venant grever le pronostic fonctionnel. De même les séquelles sensitives restent fréquentes et parfois à l’origine d’un handicap fonctionnel sévère.
Atteinte du système nerveux central (SNC)
L’atteinte du SNC se rencontre chez 5 % des patients, habituellement de façon précoce dans le cours évolutif de la maladie (11,36). Les manifestations cliniques traduisent la vascularite cérébrale : accident vasculaire avec déficit sensitif et/ou moteur (52 %), hémorragie cérébrale ou méningée (24 %) parfois due à une vascularite localisée des plexus choroïdiens, baisse d’acuité visuelle (33 %) par neuropathie optique, occlusion de l’artère centrale de la rétine plus souvent que par cécité corticale, voire troubles cognitifs, épilepsie (36). Les atteintes sont souvent intriquées.
La tomodensitométrie peut révéler des infarctus. L’imagerie par résonance magnétique est l’examen de choix. Elle objective des hypersignaux de la substance corticale et sous-corticale sur les images pondérées en T2.
Lorsqu’une angiographie est réalisée, les vaisseaux peuvent présenter des irrégularités de calibre très évocatrices de vascularite. Les séquelles sont fréquentes.
Manifestations cutanées
L’atteinte cutanée est présente dans 40 à 70 % des cas (4,9,11,37). Elle traduit la vascularite des vaisseaux de petit calibre ou des granulomes extravasculaires. Les manifestations vasculaires cutanées sont révélatrices de la GEPA dans moins de 25 % des cas (9). Un purpura vasculaire est l’atteinte cutanée la plus fréquente et s’observe dans 31 à 50 % des cas (4,9). Des nodules surviennent dans presque le tiers des cas (38). Rouges ou violacés, ils prédominent au doigt, sur le crâne et à la face d’extension des coudes ou des avant-bras. Ils sont souvent bilatéraux et symétriques. Les biopsies objectivent les granulomes extravasculaires qui ne sont pas spécifiques de la GEPA (38). D’autres manifestations cutanées peuvent survenir : syndrome de Raynaud, lésions urticariennes (9 %), livedo reticularis (6 %), papules infiltrées, vésicules ou bulles voire une gangrène d’un doigt ou d’un orteil (9).
Manifestations gastro-intestinales
Souvent redoutables, elles surviennent chez 23 % des patients (9). Les douleurs abdominales sont alors le premier symptôme dans 30 à 60 % des cas (4,9). Des nausées, des vomissements ou une diarrhée sont moins fréquents. Un méléna ou une hématémèse est présent respectivement chez 6 et 3 % des patients (9). Une perforation digestive représente la plus sévère des manifestations digestives et une cause importante de mortalité (4,9). Les manifestations gastro-intestinales représentent ainsi un des facteurs pronostiques péjoratifs de la GEPA (22,39). Elles sont incluses dans le score pronostique FFS (Five Factor Score) (Tableau 3).
L’intensité des douleurs abdominales peut suggérer une péritonite par perforation d’un organe creux ou une ischémie mésentérique qui nécessitent une intervention chirurgicale. Dans notre expérience, cette situation s’est présentée seulement 4 fois (9) : une ischémie mésentérique a été trouvée dans deux observations tandis qu’une appendicite et une colite ischémique une fois chacune.
Une vascularite et un granulome peuvent siéger tout le long du tube digestif avec une prédominance à l’intestin grêle et/ou au colon. La présence de granulomes extravasculaires de la muqueuse digestive prend parfois un aspect de pseudo-polypes (40). La présence de plusieurs ulcérations duodénales et jéjunales peut faire évoquer le diagnostic de vascularite.
L’examen anatomo-pathologique des prélèvements du tube digestif peut mettre en évidence la vascularite et/ou l’infiltration par les éosinophiles. La GEPA est une cause d’eosinophilic gastrointestinal disease (EGID) (41). Lorsque ces anomalies prédominent au niveau de la muqueuse, la traduction peut en être une anémie, une hypoalbuminémie et/ou une stéatorrhée.
L'angiographie détecte, beaucoup plus rarement que dans la PAN, la présence de sténoses et de microanévrysmes des vaisseaux.
Manifestations cardiaques
L’atteinte cardiaque est une des manifestations les plus sévères de la GEPA. Elle représente la première cause de mortalité. Sa fréquence varie entre 15 et 85 % des études (1,4,11,25,27,42). Une atteinte myocardique ou une péricardite sont survenues respectivement chez 17 et 15 % de nos patients (11). Une tamponnade ne complique que rarement la péricardite. L’endocarde est habituellement respecté. Contrairement au syndrome hyperéosinophilique où la fibrose endomyocardique est classique, celle-ci est exceptionnellement rapportée au cours de la GEPA malgré l’hyperéosinophilie que ces deux entités partagent. L’atteinte cardiaque de la GEPA fait intervenir différents mécanismes : une vascularite des branches des artères coronaires, des granulomes extravasculaires et des infiltrats interstitiels composés d’éosinophiles. Dans les séries de patients atteints de GEPA, l’infiltration éosinophilique représentait la donnée histologique la plus fréquente même si des granulomes épicardiques et myocardiques étaient également notés (1). Une vascularite nécrosante a été mise en évidence dans le péricarde chez deux patients à l’occasion de biopsie péricardique (9,43).
Le tableau clinique est dominé par l’insuffisance cardiaque qui se développe rapidement et est souvent sévère. Celle-ci a été à l’origine du décès de 3 des 11 patients de la série originale de GEPA (1) et de 14 (31 %) des 45 décès dans notre série (11). L’angor et l’infarctus du myocarde sont rares malgré la fréquence de la vascularite coronarienne (44). Elle peut régresser sous traitement (45).
L’ECG peut enregistrer les anomalies en rapport avec l’ischémie ou la cardiomyopathie. Lorsqu’une coronarographie est réalisée, des sténoses, microanévrysmes ou thromboses ne sont que rarement observées en raison de la prédominance de l’atteinte des petits vaisseaux (45). Néanmoins, les angiographies peuvent être anormales et refléter l’extension possible de la vascularite aux vaisseaux de moyen calibre (44,46). Dans le cadre d'une myocardiopathie, les biopsies endomyocardiques ne conduisent pas habituellement au diagnostic de GEPA. L’échocardiographie objective une diminution de la contractilité non spécifique de vascularite. Chez les patients asymptomatiques sur le plan cardiaque, l’IRM dépiste très souvent des anomalies myocardiques mais qui ne sont pas corrélées à l’apparition de manifestations cliniques cardiaques au cours du suivi ultérieur (47). La prise en charge ne se base donc actuellement pas sur ces seuls signes IRM qui semblent trop sensibles.
Manifestations ostéo-articulaires
Des arthralgies sont fréquentes (30 % des cas) et volontiers inaugurales. Des arthrites sont plus rares. Elles touchent volontiers les grosses articulations et sont non destructrices et non érosives.
Manifestations musculaires
Des myalgies surviennent dans 39 à 69 % des cas et régressent rapidement sous traitement (4,9,11). Parfois leur intensité peut faire évoquer une polymyosite mais les CPK sont habituellement normales (48).
Atteinte rénale
L’atteinte rénale est rare au cours de la GEPA. Dans notre série de 96 patients, elle n’a été observée que chez 22 % d’entre eux (11). Sa fréquence pourrait avoir été surestimée dans certaines études (4). Elle doit être recherchée néanmoins chez tous les patients.
Une glomérulonéphrite extracapillaire avec des croissants représente la présentation la moins rare. Dans une étude portant sur 21 patients, la glomérulonéphrite était segmentaire et focale (49). Dans certaines de ces observations, la glomérulonéphrite était rapidement progressive avec insuffisance rénale et nécessité de recourir transitoirement ou définitivement à l’hémodialyse (49). La glomérulonéphrite extracapillaire est associée à la présence d'ANCA anti-MPO.
A côté de ces glomérulonéphrites extra-capillaires, encore plus rarement sont décrites des glomérulonéphrites extra-membraneuses, notamment chez les patients n’ayant pas d’ANCA (50).
Au cours de la GEPA, il n’y a pas de vascularite rénale et l’angiographie ne montre pas de microanévrysmes. Chez les patients ayant des ANCA, il n’y a pas lieu de réaliser une angiographie avant la biopsie rénale (51). Une insuffisance rénale est fortement corrélée à une surmortalité et est incluse dans le score pronostique FFS (Tableau 3) (23,39).
Une atteinte urétérale a également été décrite au cours de la GEPA (52). La sténose concerne habituellement la partie inférieure des uretères et plus rarement la partie supérieure. Une anurie et/ou une insuffisance rénale peuvent en résulter.
Atteinte ORL
Une sinusite maxillaire est une des atteintes majeures de la GEPA et représente un des critères de classification de l’ACR. Une rhinite allergique et/ou une polypose sinusienne sont retrouvées chez 70 % des patients (4). Une histoire de sinusite chronique était retrouvée à l’interrogatoire de 63 % de nos patients atteints de GEPA (9). Il est important de différencier l’atteinte ORL de la GEPA de celle de la GPA dont la présentation peut être similaire. Dans la GEPA, il n’y a pas de destruction et le pronostic des manifestations ORL est bon, même si elles persistent après guérison de la vascularite (9,39). Après obtention d’une rémission, l’atteinte ORL évolue pour son propre compte et ne représente pas, lorsqu’elle est isolée, un signe d’activité de la vascularite.
Manifestations oculaires
Une atteinte oculaire est possible au cours de la GEPA. Des uvéites, vascularites rétiniennes, épisclérites, des nodules conjonctivaux et des pseudo-tumeurs orbitaires ont été rapportés (53,54). Une névrite optique ischémique peut aussi compliquer une GEPA. Une vascularite nécrosante des branches de l'artère temporale a été décrite (55), mais la présence de cellules géantes est exceptionnelle.
Autres manifestations
Des adénopathies périphériques ou médiastinales contenant des éosinophiles ont été décrites (4).
FORMES CLINIQUES DE LA GEPA
La GEPA a un spectre très large qui pourrait correspondre à plusieurs entités. On commence à envisager un démembrement des formes cliniques avec des mécanismes pathogéniques différents (11,56-59). La prévalence des ANCA lors du diagnostic dans ces études est de 30 à 40 % avec une fluorescence périnucléaire et une spécificité anti-myéloperoxidasique dans les ¾ des cas. Les patients dépourvus d’ANCA présentent plus volontiers des manifestations liées à l’infiltration éosinophilique : atteinte cardiaque (myocardiopathie ou péricardite éosinophilique) ou atteinte pulmonaire non-hémorragique (infiltrats, pleurésie éosinophilique). Un rôle pathogène des éosinophiles est envisagé par le bais de la libération de protéines cationiques comme au cours du syndrome hyperéosinophilique. Au contraire, le tiers des patients ayant des ANCA ont plus volontiers des manifestations directement liées à la vascularite : glomérulonéphrite, hémorragie alvéolaire, atteinte neurologique périphérique, purpura.
Ainsi, le spectre de la GEPA se précise avec d’un côté les patients avec infiltrats éosinophiliques sans vascularite ni ANCA et de l’autre ceux avec vascularite et ANCA. Néanmoins, il existe un continuum avec de nombreuses formes intermédiaires avec ou sans ANCA.
La présence d’ANCA lors du diagnostic n’affecte pas le taux de mise en rémission, ni de mortalité mais s’accompagne d’une augmentation du taux de rechute (11,60).
FORMES LOCALISÉES de la GEPA
La GEPA est une maladie systémique qui affecte plusieurs organes dans le contexte d'un asthme, d'une éosinophilie et de signes généraux. Néanmoins, à côté de cette forme habituelle systémique, des formes localisées de GEPA ont été rapportées (12). La maladie peut se résumer à l'atteinte d'un seul organe avec la présence de signes généraux. Un antécédent d'allergie est noté chez la majorité des patients même si l'asthme peut manquer lors du diagnostic. Le tube digestif et le cœur sont les organes les plus souvent touchés. Les manifestations gastro-entérologiques comprennent des cas de cholécystite éosinophilique, d'entérite éosinophilique et de vascularite mésentérique. Une insuffisance cardiaque peut également s'observer. De rares cas de nodules périphériques cutanés (61), de sténose urétérale et de manifestations cutanées ont été rapportés. L'évolution est comparable à celle de la forme systémique et dépend des organes touchés.
FACTEURS DECLENCHANTS
L'étiologie de la GEPA reste inconnue. Des facteurs favorisants très divers, qui pourraient être étiologiques dans certains cas, ont été incriminés (7,25,61,62). Jusqu'à présent, il n'a pas été possible d'identifier un antigène commun parmi tous les médicaments, particules et traitements désensibilisants suspectés. Quelque soit le facteur favorisant suspecté dans le déclenchement de la maladie ou d'une rechute, il est contre-indiqué à l'avenir. Les vaccinations avec des vaccins inactivés (grippe, pneumocoque) doivent être encouragés (63).
Plusieurs cas de GEPA ont été rapportés au cours de traitement par des antagonistes des récepteurs aux leucotriènes. Le zafirlukast et le montelukast sont les deux médicaments incriminés. Il est maintenant considéré comme très probable que l'apparition d'une GEPA soit plutôt liée à la baisse concomitante des corticoïdes rendue possible par ces thérapeutiques utilisées dans l'asthme (8) qu’aux antileucotriènes eux-mêmes (64-68). En effet, une baisse rapide ou un sevrage brutal de la corticothérapie peut aussi démasquer une GEPA sous-jacente. Quelques cas ont aussi été rapportés sous omalizumab posant la question de la responsabilité éventuelle de cet anticorps monoclonal anti-IgE (69,70). Des poussées sévères de vascularite ont également été rapportées sous ce traitement (71). Le lien de causalité reste débattu (69-71).
EXAMENS BIOLOGIQUES et COMPLÉMENTAIRES
Les caractéristiques principales sont l'éosinophilie, le syndrome inflammatoire et parfois, la positivité des ANCA.
Le syndrome inflammatoire biologique est fréquent et était noté chez 80 % de nos patients (9). L'éosinophilie est quasi-constante et habituellement supérieure à 1.500/mm3. L'éosinophilie moyenne est de 7.200 +/- 7.700 mm3 chez nos patients (9) mais elle peut atteindre 50.000/mm3 (72). Sous corticothérapie, l'éosinophilie sanguine diminue très rapidement, parfois en moins d'une semaine. Des ANCA sont détectés chez 30 à 40 % des cas par IF et/ou ELISA avec une fluorescence périnucléaire et une spécificité anti-myéloperoxidasique dans les ¾ des cas de la littérature (Figure 7) (9,11,28,30,56,58,73-78).
Le titre des ANCA n'est pas corrélé à l'évolution mais les formes avec ANCA ont une augmentation de la fréquence des rechutes (11,60). Du facteur rhumatoïde est présent chez 54 % des patients (4,27).
Lorsqu'il est réalisé, le lavage broncho-alvéolaire ramène un liquide contenant des éosinophiles.
Des anomalies radiographiques sur le cliché thoracique étaient visualisées chez 38 % de nos patients sous forme d'infiltrats bilatéraux et migrateurs et d'opacités alvéolaires de répartition inégale (9). Le scanner visualise alors des opacités en verre dépoli ainsi que des opacités parenchymateuses alvéolaires périphériques ou disséminées (Figure 10). Une sinusite peut s'observer sur le cliché radiographique mais est mieux appréciée par la tomodensitométrie.
L'angiographie peut être réalisée chez les patients avec manifestations gastro-intestinales ou en cas de doute diagnostique. Les sténoses et microanévrysmes sont moins fréquents que dans la PAN mais ont été observés chez le tiers des patients sélectionnés pour avoir cette investigation (9).
Bien que la GEPA affecte préférentiellement les petits vaisseaux, l'existence de microanévrysmes et de sténoses chez certains patients illustre l'atteinte des vaisseaux de moyen calibre et les possibilités de chevauchement entre les différents calibres des vaisseaux lésés.
EVOLUTION
Le pronostic de la GEPA a été transformé depuis l'apparition des corticoïdes et dans certains cas des immunosuppresseurs. Sous traitement, une rémission est rapidement obtenue dans plus de 80 % des cas. Une telle rémission a été observée chez 89 % de nos patients (9). Des rechutes sont survenues dans 26 % des cas, au cours de la première année pour la moitié d'entre elles et après en moyenne 69 mois d'évolution (9). Dans notre série de 338 patients analysés, les rechutes de vascularite sont survenues chez 35,2 % des patients avec ANCA versus 22,5% chez ceux sans (11). La présentation clinique de la rechute diffère des manifestations inaugurales chez la moitié des patients. Les rechutes peuvent être sévères et être à l'origine du décès. Quelques patients connaissent plusieurs rechutes s'étalant sur quelques années. Chumbley et al. ont rapporté une survie de 90 % à un an et de 62 % à 5 ans (26). Dans notre série, le taux de survie est de 79 % à 10 ans et le taux de survie sans rechute est de 54 % à 10 ans (11). La principale cause de décès est l'atteinte cardiaque, surtout par insuffisance cardiaque et beaucoup moins souvent par infarctus du myocarde. Selon la revue de la littérature effectuée par Lanham et al, la cause du décès a pu être identifiée dans 50 cas (4) : presque la moitié des patients (48 %) sont décédés d'insuffisance cardiaque ou d'infarctus du myocarde. Les autres causes de décès étaient l'insuffisance rénale (18 %), une hémorragie cérébrale (16 %), une perforation ou une hémorragie intestinale (8 %), un état de mal asthmatique (8 %) et une insuffisance respiratoire (2 %).
Il faut clairement distinguer (11,63) :
- les rechutes de la vascularite qui surviennent globalement chez 25 % des patients, surtout dans les premières années d’évolution,
- et les poussées d’asthme et/ou de sinusite, parfois accompagnées d’une augmentation de l’éosinophilie, survenant chez 19 % des patients, qui ne représentent pas isolément une rechute de la vascularite mais justifient une adaptation thérapeutique.
Dans les deux premières années d’évolution de la vascularite, à la baisse de la corticothérapie, le pourcentage de patients présentant une rechute de la vascularite (45 %) est plus important que celui présentant une exacerbation de l’asthme ou des manifestations rhinosinusiennes (22 %) (79). Par contre, ultérieurement, les décompensations d’asthme et des manifestations ORL continuent de survenir régulièrement au cours du suivi, à distance de la mise en rémission de la vascularite (80). En effet, l'asthme persiste habituellement au décours de la guérison de la vascularite et nécessite souvent une faible corticothérapie d'entretien locale ou générale.
Des séquelles neurologiques peuvent résulter d'une neuropathie périphérique déficitaire ou d'une ischémie cérébrale.
Une insuffisance cardiaque peut persister et nécessiter exceptionnellement une transplantation. Une insuffisance rénale chronique peut aussi conduire à l'épuration extrarénale définitive.
PRONOSTIC
Pour aider le clinicien à proposer la thérapeutique la plus appropriée et éviter notamment le sur-traitement au cours de la PAN, de la PAM de la GEPA, nous avons proposé en 1996 un score pronostique FFS (Five Factor Score) qui a été actualisé pour s’appliquer aussi à la GPA (Tableau 3) (23,39). Le FFS actualisé tient compte des manifestations associées à une surmortalité à savoir l’existence d’un âge supérieur à 65 ans, d'une cardiomyopathie spécifique, de manifestations gastro-intestinales, d’une insuffisance rénale définie par une créatininémie stabilisée supérieure à 150 µmol/l et de l’absence de manifestations ORL (39). Dans la GEPA, l’adaptation thérapeutique selon le FFS 1996 (23) a fait disparaître la surmortalité des formes sévères (60), validant ainsi le bien fondé de son utilisation.
TRAITEMENT
Le traitement ne se conçoit qu'en urgence et en milieu spécialisé. La corticothérapie reste la base de la thérapeutique de la GEPA, parfois associée aux immunosuppresseurs. La stratégie repose sur les recommandations de la Task Force (63) et du PNDS 2019 (81).
La corticothérapie
Les corticoïdes sont très efficaces dans la GEPA et des doses importantes doivent être initialement employées (19,63,79-82).
Les bolus de méthylprednisolone sont administrés à la posologie de 15 mg/kg en perfusion de 60 min, répétée toutes les 24 heures pendant 1 à 3 jours. Ils sont très largement utilisés à la phase initiale des vascularites systémiques sévères, particulièrement en cas de menace immédiate du pronostic vital ou à la phase d'extension d'une polyneuropathie, en raison de leur efficacité rapide et relative innocuité. Une ou plusieurs perfusions de méthylprednisolone suffit pour obtenir une amélioration rapide de l'asthme, des signes généraux ainsi que la disparition de l'éosinophilie.
Les corticoïdes (prednisone) sont donnés à la posologie d'1 mg/kg/jour. La mortalité dans notre série a diminué depuis 1996, ce qui correspond à la stratification selon le FFS (11,60). Chez les patients atteints de GEPA avec un score FFS nul, i.e. de bon pronostic, la corticothérapie seule permet d'obtenir d'emblée une rémission complète dans 77 % des cas (82). Ainsi, 84 % de ces patients atteints de GEPA de bon pronostic et traités initialement par une corticothérapie seule sont en rémission complète avec un recul de 32 +/- 19 mois (82). Les patients qui ont dû recevoir secondairement un immunosuppresseur ont connu également une évolution favorable. Avec un recul de 80 mois, que le FFS soit de 0 ou plus, la survie reste excellente mais 41 % des patients rechutent. Les rechutes surviennent en moyenne 2 ans après l’instauration du traitement, lorsque les immunosuppresseurs ont été interrompus pour la grande majorité des patients et la corticothérapie diminuée sous 10 mg/jour (60). Nous recommandons de diminuer la posologie des corticoïdes de 5 mg tous les 10 jours jusqu'à la moitié de la posologie initiale. Celle-ci est alors maintenue pendant trois semaines puis diminuée de 2,5 mg tous les 10 jours jusqu'à une posologie de 15 mg par jour. Une baisse plus progressive est alors nécessaire à raison d'une diminution d'1 mg tous les 10 jours jusqu'au sevrage. Néanmoins, il est souvent impossible d'interrompre complètement les corticoïdes en raison de l'asthme résiduel qui nécessite volontiers une corticothérapie d'entretien par voie générale (5 à 10 mg par jour) ou par inhalation. Dans les études du GFEV, 75 % des patients à 2 ans sont encore sous prednisone à une posologie moyenne inférieure à 9 mg/j (79). Seulement 15 % des patients peuvent définitivement interrompre la corticothérapie à long terme et la posologie moyenne des patients encore sous traitement à la dernière visite était de 12,9 mg/j (11).
Les immunosuppresseurs
L'indication des immunosuppresseurs, notamment du cyclophosphamide, doit reposer sur la constatation de facteurs pronostiques défavorables bien définis (23). La majorité des patients atteints de GEPA n'a pas de facteur de mauvais pronostic selon le FFS (23). Chez ces patients, le cyclophosphamide n'est pas indiqué d'emblée et ne se conçoit qu'après échec d'une corticothérapie seule ou en cas de rechute.
Le cyclophosphamide
L'indication des immunosuppresseurs, notamment du cyclophosphamide, doit reposer sur la constatation de facteurs pronostiques défavorables bien définis (23). La majorité des patients atteints de GEPA n'a pas de facteur de mauvais pronostic selon le FFS (23). Chez ces patients, le cyclophosphamide n'est pas indiqué d'emblée et ne se conçoit qu'après échec d'une corticothérapie seule ou en cas de rechute.
Le cyclophosphamide était conventionnellement administré par voie orale à la posologie de 2 mg/kg/jour. En association avec la corticothérapie, il représentait le traitement classique de la PAN. Bien que cette modalité d'administration soit efficace dans le traitement des vascularites, son rapport efficacité/toxicité est faible. Les effets indésirables les plus importants d'une telle administration quotidienne de cyclophosphamide sont la possibilité de cystite hémorragique, de cystite interstitielle, d'hypoplasie médullaire, d'insuffisance ovarienne et/ou de néoplasie (cancer de la vessie et lymphome) (83,84). Les infections représentent une cause majeure de mortalité chez les patients atteints de vascularite systémique, particulièrement s'ils reçoivent une forte corticothérapie avec des immunosuppresseurs (85). Pour tenter de diminuer la morbidité associée à l'administration quotidienne de cyclophosphamide, des protocoles ont utilisé le cyclophosphamide de façon intermittente.
Ainsi, les bolus de cyclophosphamide sont maintenant de plus en plus employés dans le traitement des vascularites nécrosantes systémiques et doivent être privilégiés à l'administration orale continue (60,81,86,87). La posologie, le nombre total et la fréquence des bolus de cyclophosphamide doivent être ajustés à l'âge du patient, à la fonction rénale, à l'hémogramme et à la réponse aux thérapeutiques antérieures. Une hydratation abondante et le mesna (Uromitexan©) doivent être associés aux bolus. L'indication du cyclophosphamide dépend du score pronostique FFS. Le cyclophosphamide ne se discute, en première intention, que chez les patients dont ce score n'est pas nul. Nous recommandons l'administration de 3 perfusions de 0,6 g/m2 de cyclophosphamide à 15 jours d’intervalle puis de 0,7 g/m2 toutes les 3 semaines jusqu’à la rémission (6 bolus en moins de 4 mois). Une fois la rémission obtenue, un traitement immunosuppresseur (azathioprine ou méthotrexate) est préconisé, par analogie aux autres vascularites pour une durée de 12 à 18 mois supplémentaires.
Le mépolizumab et les autres inhibiteurs de l’interleukine 5
L’interleukine-5 est fortement activée lors des phases d’activité de la GEPA et est particulièrement utile pour l’activation, la maturation et la survie des éosinophiles. Les inhibiteurs de l’interleukine-5 sont venus rejoindre l’arsenal thérapeutique de la granulomatose éosinophilique avec polyangéite, pour l’instant essentiellement à visée d’épargne des corticoïdes sur les manifestations respiratoires sévères en phase d’entretien. Le mepolizumab, anticorps monoclonal humanisé dirigé contre l’’interleukine-5, a l’autorisation de mise sur le marché dans l’asthme sévère éosinophilique et une autorisation de la FDA dans la GEPA. Deux études pilotes ouvertes avaient rapporté un effet d’épargne des corticoïdes du mepolizumab dans la GEPA pendant toute la phase de traitement et le maintien d’une rémission avec une éventuelle thérapeutique immunosuppressive complémentaire mais stable (88,89). Une étude internationale, randomisée, contrôlée, en double insu, de phase III, a évalué l’efficacité et la tolérance du mepolizumab par rapport au placebo, chez des patients avec essentiellement des manifestations respiratoires réfractaires ou rechutant de GEPA, malgré un traitement standard incluant une corticothérapie avec ou sans autre thérapeutique immunosuppressive (90). La durée sans recrudescence asthmatique ni poussée de vascularite était supérieure chez les patients traités par mépolizumab par rapport à ceux recevant le placebo avec une épargne cortisonée significative. Le bénéfice sur les manifestations de vascularite reste à démontrer (91). Une étude multicentrique du GFEV va évaluer l’intérêt de ce traitement en traitement d’induction de la GEPA (EMERGE).
En dehors du mepolizumab, deux autres biomédicaments à action ciblée contre l’interleukine-5 sont validés dans l’asthme éosinophilique et en cours d’investigation dans la GEPA (7). Comme le mepolizumab, le reslizumab est dirigé contre la cytokine, tandis que le benralizumab est un anticorps monoclonal humanisé dirigé contre le récepteur de l’’interleukine-5. Il empêche la fixation de l’’interleukine-5 à ses récepteurs et induit l’apoptose des éosinophiles et des basophiles.
Le rituximab
L’expérience avec le rituximab est actuellement trop faible pour pouvoir le préconiser comme alternative au cyclophosphamide en première intention du traitement d’induction des formes sévères (92-96), même si ce traitement est validé dans les vascularites sévères associées aux ANCA chez des patients atteints de GPA et de PAM. Une étude a compilé les données de 73 patients avec GEPA qui avaient reçu du rituximab, essentiellement pour une maladie réfractaire ou en rechute (95). Une efficacité du traitement d’induction par le rituximab a été observée dans la vaste majorité des observations et sur une large variété de manifestations de la maladie (7). Une série rétrospective multicentrique a analysé les données de 41 patients avec GEPA, qui ont reçu du rituximab dans 4 centres experts de prise en charge des vascularites, essentiellement pour une maladie réfractaire ou une rechute (96). Les patients ayant des ANCA ont connu un plus fort taux de mise en rémission à 12 mois : 80 % (12/15) pour ceux qui avaient des ANCA versus 38 % (8/21) pour ceux qui n’avaient pas d’ANCA détectables. Une autre étude rétrospective monocentrique de 69 patients a montré des arguments indirects en faveur d’une efficacité partielle du rituximab avec une diminution parallèle de la corticothérapie (93). Les manifestations ORL et asthmatiques semblaient peu améliorées sauf peut-être chez les patients ayant des ANCA. Dans deux observations, la survenue d’un bronchospasme sévère lors de la perfusion de rituximab a soulevé la possibilité d’une réaction d’hypersensibilité favorisée par le rituximab (94). Deux études prospectives contrôlées en double insu évaluent, sous l’égide du Groupe Français d’Etude des Vascularites, le rituximab comme traitement d’induction (REOVAS) et traitement d’entretien (MAINRITSEG) de la GEPA (www.vascularites.org).
Les anti-IgE
L’introduction des thérapeutiques anti-IgE dans l’asthme a inauguré une nouvelle ère de médicaments biologiques et s’est révélée bénéfique chez les patients avec asthme allergique sévère. Des patients avec GEPA ont aussi été traités par l’omalizumab avec des résultats variables, quelques patients ayant vu leur vascularite débuter juste après l’introduction de ce biomédicament (69,70). Comme cela est maintenant reconnu avec les antagonistes des leucotriènes, il est possible que l’omalizumab ait démasqué une vascularite sous-jacente en permettant la baisse des corticoïdes dans quelques observations. Une étude rétrospective du GFEV a inclus 17 patients atteints de GEPA ayant débuté l’omalizumab pour un asthme sévère ou une atteinte rhinosinusienne corticodépendants, alors que leur vascularite était peu ou pas évolutive (71). Le suivi médian est de 22 mois. Un effet d’épargne cortisonée semble probable mais deux de ces patients ont dû interrompre le biomédicament pour une poussée sévère d’asthme et surtout deux autres pour une névrite rétrobulbaire avec effondrement de l’acuité visuelle irréversible en rapport avec une poussée de la vascularite. Des données complémentaires sont donc nécessaires avant de pouvoir innocenter l’omalizumab dans la survenue de ces manifestations systémiques mais son efficacité semble insuffisante pour le recommander dans le traitement de la GEPA (7).
Autres traitements
Il n'y a aucun argument pour envisager des échanges plasmatiques en première intention (97).
Les gammaglobulines par voie veineuse peuvent être administrées comme dans les autres vascularites associées à la présence d'ANCA (98-100). La dose totale est de 2 g/kg en 2 perfusions (1g/kg/j ; J1 et J2) ou repartis sur 5 jours, surtout en cas d'atteinte rénale (0,4 g/kg/j ; J1 à J5). Cette thérapeutique ne peut pas être recommandée en première intention mais est parfois utile chez les patients en échec thérapeutique malgré une corticothérapie et des immunosuppresseurs, en association aux autres traitements spécifiques (vascularite réfractaire). Leur prescription doit être discutée avec un centre de compétences ou de référence, afin de pouvoir ensuite évaluer leur prescription. Le centre de référence propose aussi la mise à disposition d’informations actualisées sur les critères diagnostiques, les outils de suivi, les recommandations thérapeutiques et les protocoles en cours (www.vascularites.org).
L’azathioprine a été évaluée en double insu versus placebo, en traitement d’induction de première intention pendant 1 an, chez 51 patients avec GEPA de bon pronostic, en association à une corticothérapie conventionnelle (79). Cet immunosuppresseur n’a pas permis d’augmenter le taux de rémission prolongée sans rechutes (critère principal) ni de diminuer le taux de rechute de la vascularite (critère secondaire), la fréquence des poussées rhino-sinusiennes (critère secondaire) ou la dose cumulative de corticoïdes (analyse post-hoc). Sur le long terme, aucune différence dans le contrôle de la vascularite ou des manifestations respiratoires n’était observée chez les patients ayant reçu de l’azathioprine par rapport à ceux du bras contrôle (80).
Le méthotrexate ne semble pas être capable de diminuer les rechutes mais il a été proposé à l’induction chez une dizaine de patients avec des maladies peu sévères pour tenter d’épargner la corticothérapie (101). Les résultats de cette courte étude ouverte sont difficilement analysables en l’absence de groupe contrôle.
L'interféron alpha a été administré avec succès à des patients atteints de GEPA sévère dont certains avec insuffisance cardiaque (102,103). Néanmoins l’expérience est limitée pour l’instant avec ce traitement qui attend d'être validé sur une plus grande série de patients avant de pouvoir être recommandé.
D’autres thérapies ciblées pourraient avoir un intérêt dans la GEPA (7). Les anticorps ciblant l’interleukine-13 ont donné des résultats prometteurs dans le traitement de l’asthme, comme le tralokinumab ou le lebrikizumab. Une stratégie efficace permettant un contrôle de l’asthme est aussi possible avec un anticorps comme le dupilumab, anticorps monoclonal humanisé dirigé contre la sous-unité alpha du récepteur de l’interleukine-4 qui inhibe à la fois la signalisation de l’interleukine-4 et de l’interleukine-13. L’ampleur des résultats rapportés dans l’asthme éosinophilique ou non en font un biomédicament candidat à être évalué en priorité dans la GEPA, d’autant qu’il est aussi efficace dans les manifestations sinusiennes chroniques et la polypose nasale. Tous ces biomédicaments devront être évalués et leur bénéfice/risque apprécié dans la GEPA au cours d’études contrôlées incluant la démonstration d’un effet d’épargne cortisonée et un versant médico-économique.
L’expérience avec les anti-TNF est extrêmement limitée. Trois patients atteints de GEPA gravissime et en échec d’une corticothérapie et du cyclophosphamide ont présenté une possible réponse à l’administration transitoire d’un anti-TNF (104).
Traitements adjuvants
Les stimulations antigéniques sont à éviter.
L'ostéoporose cortisonique doit être combattue par une activité physique régulière, une alimentation riche en calcium, une supplémentation vitamino-calcique et un bisphosphonate, d'autant plus que la masse osseuse est initialement basse.
En cas d'utilisation du cyclophosphamide, une prophylaxie anti-pneumocystose est indispensable par le triméthoprime/sulfaméthoxazole en cas de lymphopénie CD4 (par exemple, Bactrim© 800/160 Forte : 1 comprimé, 3 fois par semaine ou ½ comprimé/jour).
La kinésithérapie est indiquée en cas de multinévrite déficitaire.
Enfin, le retentissement psychologique des vascularites ne doit pas être sous-estimé (105).
A RETENIR
Quand évoquer le diagnostic ?
La granulomatose éosinophilique avec polyangéite (Churg-Strauss) doit être évoqué devant tout asthme éosinophilique associé à :
- un signe extra-pulmonaire
- ou une altération de l'état général
- même en l’absence d’ANCA (absents dans 2/3 des cas)
Xavier PUÉCHAL
RÉFÉRENCES
- Churg J, Strauss L. Allergic granulomatosis, allergic angiitis and periarteritis nodosa. Am J Pathol 1951 ; 27 : 277-94.
- Masi A, Hunder G, Lie J, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis angiitis). Arthritis Rheum 1990 ; 33 : 1094-1100.
- Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013 ; 65 : 1-11.
- Lanham J, Elkon K, Pusey C, Hughes G. Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strauss syndrome. Medicine 1984 ; 63 : 65-81.
- Vaglio A, Buzio C, Zwerina J. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): state of the art. Allergy 2013 ; 68 : 261-73.
- Chaigne B, Terrier B, Thieblemont N, Witko-Sarsat V, Mouthon L. Dividing the Janus vasculitis? Pathophysiology of eosinophilic granulomatosis with polyangitis. Autoimmun Rev 2016 ; 15 : 139-45.
- Puéchal X. Therapeutic immunomodulation in eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Joint Bone Spine 2016 ; 83 : 7-10.
- Weller PF, Plaut M, Taggart V, Trontell A. The relationship of asthma therapy and Churg-Strauss syndrome: NIH workshop summary report. J Allergy Clin Immunol 2001 ; 108 : 175-83.
- Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine 1999 ; 78: 26-37.
- Sehgal M, Swanson J, DeRemee R, Colby T. Neurologic manifestations of Churg-Strauss syndrome. Mayo Clin Proc 1995 ; 70 : 337-41.
- Comarmond C, Pagnoux C, Khellaf M, Cordier JF, Hamidou M, Viallard JF, et al for the French Vasculitis Study Group. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): Clinical characteristics and long-term follow-up of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum 2013; 65: 270-81.
- Lie J. Limited forms of Churg-Strauss syndrome. Pathol Annu 1993 ; 28 : 199-220.
- Watts RA, Carruthers DM, Scott DG. Epidemiology of systemic vasculitis: changing incidence or definition? Semin Arthritis Rheum 1995 ; 25 : 28-34.
- Lane SE, Watts RA, Bentham G, Innes NJ, Scott DG. Are environmental factors important in primary systemic vasculitis? A case-control study. Arthritis Rheum 2003 ; 48 : 814-23.
- Watts RA, Lane SE, Scott DGI, Koldingsnes W, Nossent H, Gonzalez-Gay MA, Garcia-Porrua C, Bentham GA. Epidemiology of vasculitis in Europe. Ann Rheum Dis 2001 ; 60 : 1156-7.
- Fauci A, Haynes B, Katz P. The spectrum of vasculitis: clinical, pathologic, immunologic and therapeutic considerations. Ann Intern Med 1978 ; 89 : 660-76.
- Frohnert P, Sheps S. Long term follow-up study of polyarteritis nodosa. Am J Med 1967 ; 43 : 8-14.
- Guillevin L, Lê THD, Godeau P, Jais P, Wechsler B. Clinical findings and prognosis of polyarteritis nodosa and Churg-Strauss angiitis: a study in 165 patients. Br J Rheumatol 1988 ; 27 : 258-64.
- Guillevin L, Lhote F, Jarrousse B, Fain O. Treatment of polyarteritis nodosa and Churg-Strauss syndrome. A meta-analysis of 3 prospective controlled trials including 182 patients over 12 years. Ann Med Interne (Paris) 1992 ; 143 : 405-16.
- Leib E, Restivo C, Paulus H. Immunosuppressive and corticosteroid therapy of polyarteritis nodosa. Am J Med 1979 ; 67 : 941-47.
- Rose G, Spencer H. Polyarteritis nodosa. Q J Med 1957 ; 26 : 43-81.
- Guillevin L, Fain O, Lhote F, et al. Lack of superiority of steroids plus plasma exchange to steroids alone in the treatment of polyarteritis nodosa and Churg-Strauss syndrome. A prospective, randomized trial in 78 patients. Arthritis Rheum 1992 ; 35: 208-15.
- Guillevin L, Lhote F, Gayraud M, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine 1996 ; 75 : 17-28.
- Mahr A, Guillevin L, Poissonnet M, Ayme S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener's granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: a capture-recapture estimate. Arthritis Rheum 2004 ; 51 : 92-9.
- Guillevin L, Guittard T, Bletry O, Godeau P, Rosenthal P. Systemic necrotizing angiitis with asthma: causes and precipitating factors in 43 cases. Lung 1987 ; 165: 165-72.
- Chumbley L, Harrison E, DeRemee R. Allergic granulomatosis and angiitis (Churg-Strauss syndrome). Mayo Clin Proc 1977 ; 52 : 477-84.
- Haas C, Géneau C, Odinot J, et al. L'angéïte allergique avec granulome: syndrome de Churg et Strauss. Etude rétrospective de 16 observations. Ann Med Interne (Paris) 1991 ; 142 : 135-42.
- Gaskin G, Ryan J, Rees A, Pusey C. Antimyeloperoxydase antibodies in vasculitis. Relationship to ANCA and clinical diagnosis. APMIS 1990 ; 98, S19 : 33.
- Abu-Shakra M, Smythe H, Lewtas J, Badley E, Weber D, Keystone E. Outcome of polyarteritis nodosa and Churg-Strauss syndrome. Arthritis Rheum 1994 ; 37 : 1798-1803.
- Solans R, Bosch JA, Pérez-Bocanegra C, Selva A, Huguet P, Alijotas J, Orriols R, Armadans L, Vilardell M. Churg-Strauss syndome: outcome and long-term follow-up of 32 patients. Rheumatology 2001; 40: 763-71.
- Lai R, Lin S, Lee P. Churg-Strauss syndrome presenting with capillaritis and diffuse alveolar hemorrhage. Scand J Rheumatol 1998; 27: 230-2.
- Leatherman J, Davies S, Hoidal J. Alveolar hemorrhage syndromes: diffuse microvascular lung hemorrhage in immune and idiopathic disorders. Medicine 1984 ; 63 : 65-81.
- Herreman G, Ferme I, Puech H, Caubarrère I. Angéite granulomateuse de Churg et Strauss avec paralysie phrénique. Deux observations. Presse Med 1980 ; 9 : 3631.
- Cohen-Tervaert J, Kallenberg C. Neurologic manifestations of systemic vasculitides. Rheum Dis Clin North Am 1993 ; 19 : 913-40.
- Ng K, Yeung H, Loo K, Chan H, Wong C, Li P. Acute fulminant neuropathy in a patient with Churg Strauss syndrome. Postgrad Med J 1997 ; 73 : 236-8.
- André R, Cottin V, Saraux JL, Blaison G, Bienvenu B, Cathebras P, et al. Central nervous system involvement in eosinophilic granulomatosis with polyangiitis (Churg-Strauss): Report of 26 patients and review of the literature. Autoimmun Rev 2017 ; 16 : 963-9.
- Davis M, Daoud M, McEvoy M, Su D. Cutaneous manifestations of Churg Strauss syndrome: a clinicopathologic correlation. J Acad Dermatol 1997 ; 37 : 199-203.
- Finan M, Winkelmann R. The cutaneous extravascular necrotizing granuloma (Churg Strauss granuloma) and systemic disease: a review of 27 cases. Medicine 1983 ; 62: 148-58.
- Guillevin L, Pagnoux C, Seror R, Mahr A, Mouthon L, Toumelin PL for the French Vasculitis Study Group (FVSG). The Five-Factor Score Revisited: Assessment of Prognoses of Systemic Necrotizing Vasculitides Based on the French Vasculitis Study Group (FVSG) Cohort. Medicine (Baltimore). 2011; 90: 19-27.
- Suen K, Burton J. The spectrum of eosinophilic infiltration of the gastrointestinal tract and its relationship to other disorders of angiitis and granulomatosis. Hum Pathol 1979 ; 10 : 31-43.
- Lecouffe-Desprets M, Groh M, Bour B, Le Jeunne C, Puéchal X. Eosinophilic gastrointestinal disorders associated with autoimmune connective tissue disease. Joint Bone Spine 2016 ; 83 : 479-84.
- Guillevin L, Lhote F, Gallais V, Jarrousse B, Royer I, Gayraud M, Benichou J. Gastrointestinal tract involvement in polyarteritis nodosa and Churg-Strauss syndrome. Ann Med Intern 1995 ; 146 : 260-7.
- Sharma A, de Varennes B, Sniderman A. Churg-Strauss syndrome presenting with marked eosinophilia and pericardial effusion. Can J Cardiol 1993 ; 9 : 329-30.
- Kozak M, Gill E, Green L. The Churg Strauss syndrome. A case report with angiographically documented coronary involvement and a review of the literature. Chest 1995 ; 107 : 578-80.
- Isaka N, Araki S, Shibata M, al. e. Reversal of coronary artery occlusions in allergic granulomatosis and angiitis (Churg-Strauss syndrome). Am Heart J 1994 ; 128 : 609-13.
- Hasley P, Follansbee N, Couleman J. Cardiac manifestations of Churg Strauss syndrome: report of a case and review of the literature. Am Heart J 1990 ; 120 : 996-9.
- Dunogué B, Terrier B, Cohen P, Marmursztejn J, Legmann P, Mouthon L, et al. for the French Vasculitis Study Group. Impact of cardiac magnetic resonance imaging on eosinophilic granulomatosis with polyangiitis outcomes: A long-term retrospective study on 42 patients. Autoimmun Rev 2015 ;14 : 774-80.
- De Vlam K, De Keyser F, Goemaere S, Praet M, Veys E. Churg-Strauss syndrome presenting as polymyositis. Clin Exp Rheumatol 1995 ; 13 : 505-7.
- Gaskin G, Clutterbuck E, Pusey C. Renal disease in the Churg Strauss syndrome. Diagnosis, management and outcome. Contrib Nephrol 1991 ; 94 : 58-65.
- Durel CA, Sinico RA, Teixeira V, Jayne D, Belenfant X, Marchand-Adam S, for the French Vasculitis Study Group (FVSG). Renal involvement in eosinophilic granulomatosis with polyangiitis (EGPA): a multicentric retrospective study of 63 biopsy-proven cases. Rheumatology (Oxford) 2020: keaa416.
- Guillevin L, Lhote F, Brauner M, Casassus P. Antineutrophil cytoplasmic antibodies (ANCA) and abnormal angiograms in polyarteritis nodosa and Churg Strauss syndrome: indications for the diagnosis of microscopic polyangiitis. Ann Med Interne (Paris) 1995 ; 146 : 548-50.
- Azar N, Guillevin L, Huong DL, Herreman G, Meyrier A, Godeau P. Symptomatic urogenital manifestations of polyarteritis nodosa and Churg Strauss angiitis: analysis of 8 of 165 patients. J Urol 1989 ; 142 : 136-8.
- Nissim F, Von der Valde J, Czernobilsky B. A limited form of Churg-Strauss syndrome. Ocular and cutaneous manifestations. Arch Pathol Lab Med 1982 ; 106 : 305-7.
- Takanashi T, Uchida S, Arita M, Okada M, Kashii S. Orbital inflammatory pseudotumor and ischemic vasculitis in Churg-Strauss syndrome: report of two cases and review of the literature. Ophtalmology 2001 ; 108 : 1129-33.
- Généreau T, Lortholary O, Pottier MA, Michon-Pasturel U, Ponge T, de Wazieres B, et al. Temporal artery biopsy : a diagnostic tool for systemic necrotizing vasculitis. Arthritis Rheum 1999 ; 42 : 2674-81.
- Sablé-Fortassou R, Cohen P, Mahr A, Pagnoux C, Mouthon L, Jayne D, et al. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Int Med 2005 ; 143 : 632-8.
- Hoffman G, Langford C. Are they different forms of life in the antineutrophil cytoplasmic antibody universe (editorial) ? Ann Int Med 2005 ; 143 : 683-5.
- Sinico RA, Di Toma L, Maggiore U, Bottero P, Radice A, Tosoni C, et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis Rheum 2005 ; 52 : 2926-35.
- Kallenberg CG. Churg-Strauss syndrome: just one disease entity (editorial) ? Arthritis Rheum 2005 ; 52 : 2589-93.
- Samson M, Puéchal X, Devilliers H, Ribi C, Cohen P, Stern M, for the French Vasculitis Study Group. Long-term outcomes of 118 patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) enrolled in two prospective trials. J Autoimmun 2013; 43: 60-9.
- Churg A, Brallas M, Cronin S, Churg J. Formes frustes of Churg Strauss syndrome. Chest 1995 ; 108 : 320-3.
- Phanupak P, Kohler P. Onset of polyarteritis nodosa during allergic hyposensitization treatment. Am J Med 1980 ; 68 : 1866-72.
- Groh M, Pagnoux C, Baldini C, Bel E, Bottero P, Cottin V, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) (EGPA) Consensus Task Force recommendations for evaluation and management. Eur J Intern Med 2015 ; 26 : 545-53.
- Drazen J, Israel E, O'Byrne P. Treatment of asthma with drugs modifying the leukotriene pathway. N Engl J Med 1999; 340: 197-206.
- Honsinger R. Zafirlukast and Churg Strauss syndrome. JAMA 1998 ; 279 : 1949.
- Katz R, Papernik M. Zafirlukast and Churg-Strauss syndrome. JAMA 1998 ; 279 : 1949.
- Knoell D, Lucas J, Allen J. Churg Strauss syndrome associated with zafirlukast. Chest 1998 ; 114 : 332-4.
- Wechsler M, Garpestad E, Flier S, et al. Pulmonary infiltrates, eosinophilia and cardiomyopathy following corticosteroid withdrawal in patients with asthma receiving zafirlukast. JAMA 1998 ; 279 : 455-7.
- Winchester DE, Jacob A, Murphy T. Omalizumab for asthma. N Engl J Med 2006 ; 355 : 1281-2.
- Puéchal X, Rivereau P, Vinchon F. Churg Strauss syndrome associated with omalizumab. Eur J Intern Med 2008 ; 19 : 364-6.
- Jachiet M, Samson M, Cottin V, Kahn JE, Le Guenno G, Bonniaud P et al, for the French Vasculitis Study Group. Anti-IgE monoclonal antibody in refractory and relapsing eosinophilic granulomatosis with polyangiitis (Churg-Strauss): data from 17 patients.
- Hübner C, Dietz A, Stremmel W, Stiehl A, Andrassy K. Macrolide-induced Churg Strauss syndrome in a patient with atopy. Lancet 1997 ; 350 : 563.
- Guillevin L, Visser H, Noël LH, et al. Antineutrophil cytoplasm antibodies in systemic polyarteritis nodosa with and without hepatitis B virus infection and Churg Strauss syndrome--62 patients. J Rheumatol 1993 ; 20 : 1345-9.
- Cohen-Tervaert J, Goldschmeding R, Elema JD, von den Borne A, Kallenberg CG. Antimyeloperoxidase antibodies in the Churg Strauss syndrome. Thorax 1991 ; 46 : 70-1.
- Schnabel A, Hauschild S, Gross WL. Anti-neutrophil cytoplasmic antibodies in generalized autoimmune diseases. Int Arch Allergy Immunol 1996 ; 109 : 201-6.
- Reid AJ, Harrison BD, Watts RA, Watkin SW, McCann BG, Scott DG. Churg-Strauss syndrome in a district hospital. Q J Med 1998 ; 91 : 219-29.
- Della Rossa A, Baldini C, Tavoni A, Tognetti A, Neglia D, Sambuceti G, et al. Churg-Strauss syndrome: clinical and serological features of 19 patients from a single Italian centre. Rheumatology (Oxford) 2002 ; 41 : 1286-94.
- Keogh KA, Specks U. Churg-Strauss syndrome: clinical presentation, antineutrophil cytoplasmic antibodies, and leukotriene receptor antagonists. Am J Med 2003 ; 115 : 284-90.
- Puéchal X, Pagnoux C, Baron G, Quéméneur T, Néel A, Agard C, et al. Adding azathioprine to remission-induction glucocorticoids for eosinophilic granulomatosis with polyangiitis (Churg-Strauss), microscopic polyangiitis or polyarteritis nodosa without poor prognosis factors. A randomized, controlled trial. Arthritis Rheumatol 2017 ; 69 : 2175-86.
- Puéchal X, Pagnoux C, Baron G, Lifermann F, Geffray L, Quéméneur T, et al for the French Vasculitis Study Group investigators. Non-severe eosinophilic granulomatosis with polyangiitis: Long-term outcomes after remission-induction trial. Rheumatology 2019 ; 58 : 2107-16.
- Protocole National de Diagnostic et de Soins 2019. Vascularites nécrosantes systémiques (périartérite noueuse et vascularites associées aux ANCA). https://www.has-sante.fr/jcms/p_3076472/fr/vascularites-necrosantes-systemiques-periarterite-noueuse-et-vascularites-associees-aux-anca.
- Ribi C, Cohen P, Pagnoux C, Mahr A, Arène JP, Lauque D, et al for the French Vasculitis Study Group. Treatment of Churg-Strauss syndrome without poor-prognosis factors: a multicenter, prospective, randomized, open-label study of 72 patients. Arthritis Rheum 2008 ; 58 : 586-94.
- Stillwell T, Benson R. Cyclophosphamide-induced hemorrhagic cystitis: a review of 100 patients. Cancer 1988 ; 61 : 451-7.
- Baker G, Kahl L, Zee B, Stolzer B, Agarwal A, Medsger TJ. Malignancy following treatment of rheumatoid arthritis with cyclophosphamide. Long-term case-control follow-up study. Am J Med 1987 ; 83 : 1-9.
- Jarrousse B, Guillevin L, Bindi E, et al. Increased risk of Pneumocystis carinii pneumonia in patients with Wegener's granulomatosis. Clin Exp Rheumatol 1993 ; 11 : 615-21.
- Gayraud M, Guillevin L, Cohen P, et al. Treatment of good-prognosis polyarteritis nodosa and Churg-Strauss syndrome: comparison of steroids and oral or pulse cyclophosphamide in 25 patients. Br J Rheumatol 1997 ; 36 : 1290-7.
- Cohen P, Pagnoux C, Mahr A, Arène JP, Mouthon L, Le Guern V, et al. Churg-Strauss syndrome with poor-prognosis factors: A prospective multicenter trial comparing glucocorticoids and six or twelve cyclophosphamide pulses in forty-eight patients. Arthritis Rheum 2007 15 ; 57 : 686-93.
- Kim S, Marigowda G, Oren E, Israel E, Wechsler ME. Mepolizumab as a steroid-sparing treatment option in patients with Churg-Strauss syndrome. J Allergy Clin Immunol 2010;125:1336-43.
- Moosig F, Gross WL, Herrmann K, Bremer JP, Hellmich B. Targeting interleukin-5 in refractory and relapsing Churg-Strauss syndrome. Ann Intern Med 2011;155:341-3.
- Wechsler ME, Akuthota P, Jayne D, Khoury P, Klion A, Langford CA, et al. Mepolizumab or placebo for eosinophilic granulomatosis with polyangiitis. N Engl J Med 2017 ; 376 : 1921-32.
- Guillevin L. Vasculitis: Mepolizumab for eosinophilic granulomatosis with polyangiitis. Nat Rev Rheumatol 2017 ; 13 : 518-9.
- Thiel J, Hässler F, Salzer U, Voll RE, Venhoff N. Rituximab in the treatment of refractory or relapsing eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome). Arthritis Research & Therapy 2013, 15:R133.
- Teixeira V, Mohammad AJ, Jones RB, Smith R, Jayne D. Efficacy and safety of rituximab in the treatment of eosinophilic granulomatosis with polyangiitis. RMD Open 2019 ; 5 : e000905.
- Bouldouyre MA, Cohen P, Guillevin L. Severe bronchospasm associated with rituximab for refractory Churg-Strauss syndrome. Ann Rheum Dis 2009 ; 68 : 606.
- Fanouriakis A, Kougkas N, Vassilopoulos D, Fragouli E, Repa A, Sidiropoulos P. Rituximab for eosinophilic granulomatosis with polyangiitis with severe vasculitic neuropathy: Case report and review of current clinical evidence. Semin Arthritis Rheum 2015 ; 45 : 60-6.
- Mohammad AJ, Hot A, Arndt F, Moosig F, Guerry MJ, Amudala N, et al. Rituximab for the treatment of eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Ann Rheum Dis 2016 ; 75 : 396-401.
- Guillevin L, Lhote F, Cohen P, et al. Corticosteroids plus pulse cyclophosphamide and plasma exchanges versus corticosteroids plus pulse cyclophosphamide alone in the treatment of polyarteritis nodosa and Churg-Strauss syndrome patients with factors predicting
- Hamilos D, Christensen J. Treatment of Churg Strauss syndrome with high-dose intravenous immunoglobulins. J Allergy Clin Immunol 1991 ; 88 : 823-4.
- Jayne DR, Lockwood CM. Intravenous immunoglobulin as sole therapy for systemic vasculitis. Br J Rheumatol 1996 ; 35 : 1150-3.
- Danieli MG, Cappelli M, Malcangi G, Logullo F, Salvi A, Danieli G. Long term effectiveness of intravenous immunoglobulin in Churg-Strauss syndrome. Ann Rheum Dis 2004 ; 63 : 1649-54.
- Metzler C, Hellmich B, Gause A, Gross WL, de Groot K. Churg Strauss syndrome; successful induction of remission with methotrexate and unexpected high cardiac and pulmonary relapse ratio during maintenance treatment. Clin Exp Rheumatol 2004 ; 22 (6 Suppl 36) : S52-61.
- Tatsis E, Schnabel A, Gross W. Interferon alpha treatment in four patients with the Churg Strauss syndrome. Ann Intern Med 1998 ; 129 : 370-4.
- Metzler C, Schnabel A, Gross WL, Hellmich B. A phase II study of interferon-alpha for the treatment of refractory Churg-Strauss syndrome. Clin Exp Rheumatol 2008 ; 26 : S35-40.
- Arbach O, Gross WL, Gause A. Treatment of refractory Churg-Strauss-Syndrome (CSS) by TNF-alpha blockade. Immunobiology 2002 ; 206 : 496-501.
- Koutantji M, Pearce S, Harrold E. Psychological aspects of vasculitis. Rheumatology 2000 ; 39: 1173-9.
Figure 1. Nomenclature révisée de la Conférence de Chapel Hill 2012

Figures 4. Nomenclature révisée de la Conférence de Chapel Hill 2012
Figure 7. ANCA
Figure 9. Physiopathie de la GEPA selon Vaglio A. Allergy 2013
Figure 10. Infiltrat pulmonaire labile en verre dépoli
CLICHE 6 hémorragie alvéolaire
CLICHE 7 hémorragie alvéolaire
Tableau 1. Critères de classification de la GEPA selon l'ACR
Tableau 2. Manifestations cliniques de la GEPA
Tableau 3. FFS Facteurs pronostiques péjoratifs de la GEPA




