



Présentation et Classification
Définition et classification des Vascularites
Définition
Sous le terme de vascularites systémiques, on désigne un groupe d'affections caractérisées par une atteinte inflammatoire des vaisseaux sanguins artériels, capillaires et veineux conduisant à une altération de la paroi vasculaire, qui intéresse aussi bien l'endothélium que la média ou l'adventice. Les sténoses ou l'occlusion des lumières vasculaires par une thrombose ou une prolifération intimale sont la conséquence de l'atteinte de l'endothélium vasculaire.
Les vascularites systémiques sont parfois graves et engagent le pronostic vital. Morbidité et mortalité sont la conséquence de manifestations multiviscérales, de localisations particulières ou de complications des traitements. Une meilleure connaissance de l'étiologie, des mécanismes immunopathologiques et de l'histoire naturelle de ces affections permet d'optimiser l'approche thérapeutique et de l'adapter à chaque type de vascularite.
Classification
La plupart des classifications prennent en compte des critères cliniques et histologiques. Les critères histologiques sont :
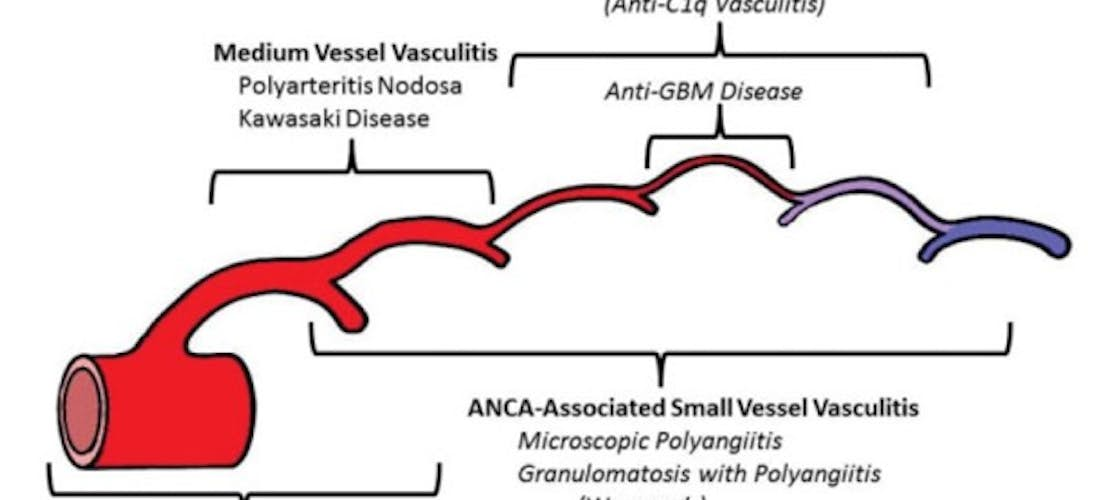
– le type et le calibre des vaisseaux atteints (gros vaisseaux : aorte et ses branches de divisions ; vaisseaux dits de petit calibre : capillaires et vaisseaux pré- et post-capillaires ; vaisseaux de moyen calibre occupant une position intermédiaire) (figure1);
– le type de l'atteinte vasculaire (nature de l'infiltrat inflammatoire, présence d'une nécrose fibrinoïde de la paroi vasculaire ou d'un granulome extravasculaire).
En 1990, l'American College of Rheumatology (ACR) a établi une classification des principales vascularites systémiques (77,78,79) fondée sur des critéres cliniques, biologiques et histologiques qui ne doivent toutefois pas être utilisés comme critères diagnostiques. Nous mentionnerons sous forme de tableaux certains des critères de classification des principales vascularites nécrosantes.
En 1994, la Nomenclature de Chapel Hill (104)(tableau 1) a permis de mieux classer la plupart des vascularites et de mettre en perspective les critères histologiques et les mécanismes pathogéniques.
Principales Vascularites Systémiques
Maladie de Takayasu
C'est la plus fréquente des artériopathies inflammatoires du sujet jeune. C'est une aorto-artérite non spécifique touchant l'aorte, les artères qui en naissent et les artères pulmonaires. L'atteinte de la crosse aortique est responsable de la rétinopathie ischémique, des complications neurologiques et de l'atteinte axillo-sous-clavière qui est classiquement à l'origine de l'abolition des pouls aux membres supérieurs. L'atteinte de l'aorte thoraco-abdominale se traduit, le plus souvent, par une hypertension rénovasculaire par sténose uni- ou bilatérale des artères rénales. Cette vascularite n'est pas nécrosante et la paroi vasculaire est le siège de cellules géantes. Elle est classée en formes distinctes selon la topographie des atteintes vasculaires
Artérite à cellules géantes (maladie de Horton)
Cette vascularite, particulière par sa topographie, atteint préférentiellement les artères de gros et moyen calibre, principalement du territoire céphalique. Elle elle peut être plus diffuse. Histologiquement, l'atteinte vasculaire intéresse les trois tuniques, avec un infiltrat inflammatoire essentiellement mononuclée, une destruction du tissu élastique et une réaction histiocytaire à son contact. On constate un épaississement intimal constitué d'une prolifération fibroblastique, et des cellules géantes peuvent être observées au contact de la limitante élastique interne (figure 3). La maladie de Horton survient dans la majorité des cas chez les sujets âgés. Sa présentation clinique, très polymorphe, impose de pratiquer, comme pour toute vascularite, une biopsie afin d'en affirmer le diagnostic.
Périartérite noueuse
C'est une vascularite inflammatoire qui touche les artères de petit et moyen calibre, avec des lésions segmentaires et transmurales siégeant volontiers aux bifurcations artérielles. L'architecture normale de la paroi vasculaire est détruite (figure 4), et la lésion peut être le siège d'une dilatation anévrismale, pouvant être objectivée par une artériographie, ou d'une thrombose (figure 5). L'American College of Rheumatology a établi des critères de classification (77) qui, malheureusement, ne permettent pas de la distinguer de la polyangéite microscopique . La conférence internationale de Consensus de Chapel Hill (104), a proposé une nomenclature plus homogène. Les vascularites sont classées en fonction de la taille des vaisseaux : vascularites non nécrosantes des gros vaisseaux (artérite de Takayasu et artérite à cellules géantes), vascularites des artères de moyen calibre (périartérite noueuse ou maladie de Kussmaul-Maier et artérite de Kawasaki), vascularite des vaisseaux de petit calibre, comportant les artérites associées aux ANCA (polyangéite microscopique, angéite granulomateuse de Churg-Strauss et granulomatose de Wegener). Les vascularites des maladies autoimmunes, les vascularites des cryoglobulinémies et le purpura rhumatoide, font partie de ce groupe.
Maladie de Kawasaki
Il s'agit d'une vascularite des artères de gros et moyen calibre, d'étiologie inconnue, qui touche préférentiellement le nourrisson et l'enfant de moins de 5 ans, plus rarement l'adulte. Le syndrome de Kawasaki réalise un syndrome adéno-cutanéo-muqueux fébrile. Sa gravité est liée à l'atteinte cardiaque et au développement d'anévrismes coronaires qui peuvent être à l'origine de mort subite.
Vascularites associées aux ANCA
- Polyangéite microscopique
C'est une vascularite des petits vaisseaux, artérioles, capillaires et veinules, sans granulome extravasculaire. La capillarite est responsable d'une glomérulonéphrite nécrosante segmentaire et focale, associée à une prolifération extracapillaire. S'y associent d'autres atteintes viscérales touchant surtout la peau, les muscles, les articulations, le poumon (hémorragie alvéolaire) et l'appareil digestif. Une certaine confusion a longtemps régné entre PAN et polyangéite microscopique, alors que ces deux maladies sont distinctes et ont des mécanismes pathogéniques différents, la polyangéite étant associée aux anticorps anticytoplasme des polynucléaires neutrophiles (ANCA). Nous résumons au tableau 2 les principales différences entre les deux maladies.
- Granulomateuse éosinophilique avec polyangéite (Churg et Strauss)
Elle se caractérise, cliniquement, par l'existence d'un asthme grave, d'une hyperéosinophilie sanguine et d'une angéite nécrosante, cliniquement proche de la PAN, touchant les artères et les veinules de petit calibre. Des infiltrats à éosinophiles et des granulomes gigantocellulaires, périvasculaires et surtout extravasculaires s'associent aux lésions vasculaires. Les trois éléments histologiques (nécrose fibrinoïde de la paroi des vaisseaux de petit calibre, infiltrats tissulaires à éosinophiles et granulomes extravasculaires), caractéristiques de l'affection, ne coexistent pas toujours sur le même site biopsique. Les critéres de classification de l'ACR sont énoncés au tableau 3.
- Granulomatose avec polyangéite (de Wegener)
Il s'agit d'une vascularite systémique dont les lésions siègent préférentiellement au niveau des voies aériennes supérieures, du poumon et des reins. La triade histologique classique de la GW associe des granulomes des voies aériennes supérieures et/ou inférieures, une vascularite nécrosante ou granulomateuse des artères de petit calibre et des veines, et une glomérulonéphrite nécrosante segmentaire et focale. Une glomérulonéphrite nécrosante segmentaire et focale, associée à une prolifération extracapillaire, est l'atteinte rénale la plus fréquente et la plus typique. Les ANCA sont présents dans environ 90 p. 100 des GW actives, il s'agit typiquement de c-ANCA ayant une spécificité anti-PR3. Les critères de classification de l'ACR sont mentionnés au tableau 4(79).
Angéite d'hypersensibilité
Ce terme est synonyme de vascularite des petits vaisseaux, de vascularite allergique et de vascularite leucocytoclasique. Il correspond à un groupe vaste et hétérogène de maladies, qui ont en commun l'atteinte des vaisseaux artériels et veineux de petit calibre, des veinules et des artérioles habituellement indemnes au cours de la PAN. Contrairement à la PAN, toutes les lésions histologiques ont le même âge. La plupart des angéites d'hypersensibilité répondent à la suppression de la substance antigénique responsable, le plus souvent un médicament.
Vascularite à IgA (Purpura rhumatoïde de Schönlein-Henoch)
Cette vascularite est le plus souvent présente chez l'enfant, touchant essentiellement la peau, le tube digestif, les articulations et les reins. La présentation clinique habituelle est un purpura vasculaire infiltré, siégeant aux membres inférieurs, parfois aux mains et à la face, favorisé par l'orthostatisme associé à des arthralgies et des douleurs abdominales (81). Histologiquement, le purpura rhumatoïde est caractérisé par une vascularite aiguë des artérioles et des veinules dans le derme superficiel et l'intestin. En immunofluorescence, il existe des dépôts d'IgA dans la paroi des artérioles et des glomérules rénaux. Le pronostic, habituellement bon, dépend de la gravité de l'atteinte rénale (néphropathie glomérulaire à dépôts mésangiaux d'IgA) et de la sévérité des atteintes digestives. Ces dernières sont habituellement bénignes chez l'enfant mais sont la première cause de mortalité chez l'adulte.
Maladie de Buerger
Appelée aussi thrombo-angéite oblitérante, c'est une vascularite des hommes jeunes, tabagiques, touchant principalement les artères et les veines de moyen et petit calibre des quatre membres, exceptionnellement les vaisseaux cérébraux et viscéraux. À l'artériographie, l'atteinte des artères sous-poplitées est presque constante, et les lésions sont strictement sous-poplitées dans plus de 80des cas.
Cryoglobulines
Ces immunoglobulines sériques précipitent à des températures inférieures à 37°C et sont associées à une vascularite caractérisée, histologiquement, par une nécrose fibrinoïde de la paroi des petits vaisseaux, avec un infiltrat inflammatoire à prédominance de polynucléaires neutrophiles dont certains peuvent être pycnotiques (leucocytoclasie). Il existe des dépôts hyalins intravasculaires avec, en immunofluorescence, un dépôt d'immunoglobulines dont la composition est celle du cryoprécipité. On distingue trois types de cryoglobulines. Les cryoglobulines de type I sont composées d'immunoglobulines monoclonales, le plus souvent une IgM, plus rarement une IgG ; elles s'observent au cours des hémopathies lymphoïdes. Dans 75 p. 100 des cas, les cryoglobulines sont mixtes, composées d'au moins deux variétés d'immunoglobulines, Les cryoglobulines mixtes de type II, avec un composant monoclonal, sont le plus souvent IgM-IgG ; le constituant monoclonal est l'IgM possédant une activité anti-IgG. Les cryoglobulines mixtes de type III ne contiennent aucun constituant monoclonal et sont habituellement composées d'IgM et d'IgG. Elles s'observent au cours des affections auto-immunes (lupus, polymyosites, PAN), de nombreuses maladies infectieuses, notamment au cours de l'hépatite C qui est retrouvée dans plus de 50des cas.
Vascularites associées aux cancers et hémopathies
Elles représentent 3 à 8 p. 100 des vascularites. Bien que le caractère paranéoplasique de ces vascularites ne puisse toujours être affirmé, l'évolution parallèle des deux pathologies suggère un lien de causalité. La survenue de la vascularite peut parfois précéder la découverte du cancer de plusieurs mois. Plusieurs types de vascularite peuvent se rencontrer, les plus fréquentes étant les vascularites leucocytoclasiques ; des vascularites du type de la PAN, granulomateuses ou proches du purpura rhumatoïde ont été aussi rapportées. Les manifestations cutanées et la fièvre sont les symptômes les plus courants, les atteintes articulaires ou neurologiques étant plus rares. Les hémopathies associées aux vascularites sont principalement les leucémies à tricholeucocytes et les myélodysplasies, ainsi que les lymphomes malins, hodgkiniens ou non. Quand aux tumeurs solides, elles sont préférentiellement bronchiques, colique ou rénales. L'évolution de la vascularite est généralement marquée par une corticosensibilité et une autonomie vis-à-vis de la néoplasie sous-jacente. Le mécanisme de ces vascularites est inconnu.
Apport des anticorps anti-cytoplasme des polynucléaires
Les anticorps anti-cytoplasme des polynucléaires neutrophiles (ANCA) sont une nouvelle famille d'auto-anticorps dirigés contre des antigènes du cytoplasme des polynucléaires neutrophiles et des monocytes. Ils ont une forte spécificité pour le groupe des vascularites nécrosantes systémiques.
Les ANCA sont retrouvés dans un groupe limité de vascularites, qui sont toutes des vascularites des vaisseaux de petit calibre. Il s'agit de la granulomatose de Wegener, de la polyangéite microscopique et de l'angéite granulomateuse de Churg et Strauss. Bien que des ANCA ont été décrits au cours d'autres affections (rectocolite ulcéro-hémorragique, maladie de Crohn, cholangite sclérosante primitive, cirrhose biliaire primitive), leur spécificité pour le groupe des vascularites nécrosantes systémiques et/ou des glomérulonéphrites rapidement progressives pauci-immunes est très élevée, de l'ordre de 94 à 99 p. 100.
Détection des ANCA
Le tableau 5 présente succinctement les principaux types d'ANCA. La méthode de référence pour la détection des ANCA est un test d'immunofluorescence indirecte sur des polynucléaires fixés dans l'alcool. Deux types de fluorescence sont observés :
- une fluorescence cytoplasmique des polynucléaires neutrophiles et des monocytes, appelée c-ANCA ;
- une fluorescence périnucléaire des polynucléaires, appelée p-ANCA.
Lorsque l'aspect en immunofluorescence ne correspond ni à celui des c-ANCA, ni à celui des p-ANCA, il est par convention atypique.
Parallèlement à l'identification des antigènes cibles des ANCA, des tests par ELISA ont été développés. Le plus souvent, les ANCA sont des anticorps de classe IgG. Des IgM ANCA ont été décrits et seraient associés, avec une fréquence élevée, à des hémorragies alvéolaires sévères ; des IgA ANCA ont été identifiés chez des patients atteints de purpura rhumatoïde de Schönlein-Henoch. Habituellement, les patients ont soit des c-ANCA, soit des p-ANCA, leur association est cependant possible chez un même sujet.
Antigènes reconnus par les ANCA
Plusieurs enzymes lysosomiales, contenues dans les granulations primaires et secondaires des polynucléaires neutrophiles, ont été identifiées comme les cibles des ANCA. Les deux principaux antigènes connus sont la PR3 (protéinase 3) et la MPO (myéloperoxydase), contenues dans les granulations primaires des polynucléaires neutrophiles et des monocytes. La PR3 est l'antigène reconnu par la majorité des c-ANCA, la MPO l'antigène reconnu par la majorité des p-ANCA (80 à 90 p. 100). Un faible pourcentage de p-ANCA peut réagir avec d'autres constituants des granules primaires (élastase, cathepsine G) ou des granules secondaires (lactoferrine).
Un certain nombre d'études ont démontré la corrélation du titre d'ANCA à l'activité clinique de la maladie et une ascension des taux d'anticorps précéde habituellement la rechute clinique. Le parallélisme entre l'activité de la GW et le titre d'ANCA n'est cependant pas total. En outre, d'authentiques GW disséminées ne s'accompagnent pas d'ANCA. Certains patients en rémission gardent des taux élevés d'ANCA, sans qu'une rechute soit observée.
Conclusion
La classification des vascularites permet de mieux reconnaître des maladies qui sont bien souvent très éloignées les unes des autres. L'impact des classifications est majeur dans la mesure où l'évolution et le pronostic sont différents et où les traitements doivent être adaptés à chaque situation clinique. La classification qui paraît avoir le plus grand intérêt aujourd'hui est celle définie par la nomenclature de Chapel Hill en raison de sa simplicité de de son édéquation avec les mécanismes pathogéniques de ces maladies. Nous soulignerons toutefois qu'aucune classification n'est parfaite et que les formes de chevauchement et les atypies restent nombreuses.




