


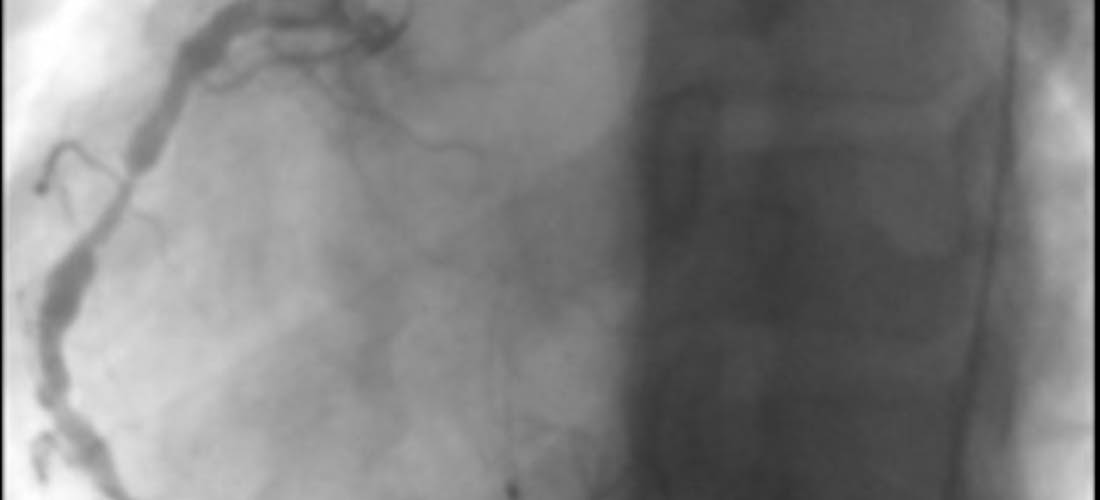

Maladie de Kawasaki
Il s'agit d'une vascularite des artères de gros et moyen calibre, d'étiologie inconnue, qui touche préférentiellement le nourrisson et l'enfant de moins de 5 ans, plus rarement l'adulte. Le syndrome de Kawasaki réalise un syndrome adéno-cutanéo-muqueux fébrile. Sa gravité est liée à l'atteinte cardiaque et au développement d'anévrismes coronaires qui peuvent être à l'origine de mort subite.
INTRODUCTION
La maladie de Kawasaki (MK) est une vascularite systémique aiguë des artères de petit et moyen calibre, d'étiologie inconnue et de début brutal, qui atteint préférentiellement les patients âgés de 6 mois à 5 ans et exceptionnellement les adultes. Elle a été décrite pour la première fois en 1967 par le pédiatre japonais Tomisaku Kawasaki. La présentation typique associe une fièvre de plus de 5 jours et des anomalies cutanéo-muqueuses.
La MK est particulièrement fréquente au Japon, où l’incidence est comprise entre 112 et 223 pour 100 000 enfants âgés de moins de 5 ans, comparativement aux Etats-Unis, où elle est d’environ 20 pour 100 000 enfants âgés de moins de 5 ans. L’incidence est 2,5 fois plus élevée chez les patients d’origine asiatique que chez les patients d’origine caucasienne, ce qui suggère une prédisposition génétique. La maladie est endémique dans les pays de l’hémisphère Nord avec des pics d’incidence d’allure épidémique ainsi qu’une recrudescence hivernale des cas, suggérant une composante infectieuse qui n’a pas encore été identifiée à ce jour.
ETIOLOGIES
Bien que l’étiologie de la MK ne soit pas encore connue, l’implication d’un ou de plusieurs agents infectieux ubiquitaires sur un terrain génétique prédisposant est fortement suspectée.
HISTOLOGIE
La MK est une vascularite systémique touchant l’ensemble des artères de petit et moyen calibre avec un tropisme particulier pour les artères coronaires. En phase aiguë, les lésions prédominent au niveau de la media avec une dissociation œdémateuse des cellules musculaires lisses et un œdème de l’intima. L’infiltrat, initialement à prédominance de neutrophiles, devient rapidement riche en cellules mononuclées et en plasmocytes à IgA. Une destruction de la limitante élastique et une prolifération fibroblastique survient ensuite, laissant place au remodelage vasculaire.
DIAGNOSTIC
En l’absence de caractéristiques cliniques pathognomoniques et de test diagnostique unique spécifique, le diagnostic de MK repose sur une combinaison de critères cliniques. On distingue une forme classique et des formes incomplètes, de diagnostic plus difficile, pour lesquelles les données biologiques et/ou échographiques peuvent aider au diagnostic.
Le diagnostic de la forme classique ou typique repose sur l’association d’une fièvre évoluant depuis au moins 5 jours et d’au minimum 4 des 5 critères suivants : 1) modifications des lèvres et de la cavité buccale (chéilite érythémateuse et/ou fissurée, langue framboisée, stomatite et pharyngite diffuses) (96%), 2) exanthème très polymorphe (96%), 3) conjonctivite bilatérale non purulente (89%), 4) anomalies des extrémités (œdème et/ou érythème des mains et des pieds au stade aigu, desquamation du bout des doigts/orteils habituellement 2 à 3 semaines après le début de la fièvre) (76%), 5) adénopathies cervicales souvent unilatérales (dont une >1,5 cm) (63%).
Les principaux diagnostics différentiels sont :
|
|
|
|
|
En l’absence de traitement, la fièvre est résolutive en 11 jours en moyenne mais peut durer jusqu’à 4 semaines (voire plus). Les différents signes cliniques apparaissent souvent de façon séquentielle et il n’est pas nécessaire qu’ils soient présents de façon concomitante pour être retenus comme des critères de MK.
Biologiquement, il existe de façon quasi-constante un syndrome inflammatoire avec élévation de la protéine C-réactive (CRP), le plus souvent au-dessus de 30 mg/l, de la procalcitonine et de la vitesse de sédimentation. Une hyperleucocytose >15 000/mm3 est fréquente mais une leucopénie est également possible. Une anémie inflammatoire modérée peut être présente et on observe parfois des anémies hémolytiques sévères après IgIV, du fait de la transmission passive d’IgG anti-A ou anti-B chez des patients de groupe A, B ou AB. Une thrombopénie à la phase aiguë peut être le reflet d’un syndrome d’activation macrophagique ou d’une coagulation intra-vasculaire disséminée (CIVD), tandis qu’une thrombocytose >450 000/mm3 est très fréquemment observée à partir de la 2ème semaine de fièvre. L’absence d’élévation de la VS, de la CRP et du chiffre des plaquettes à 7 jours de fièvre rend d’ailleurs peu probable le diagnostic de MK.
La MK de l’adulte est particulièrement rare, expliquant un délai diagnostique estimé à 13 jours en médiane. Dans la revue de littérature de Fraison et al., il a été retrouvé une fréquence similaire des principaux signes de la maladie entre les enfants et les adultes, en dehors d’anomalies des extrémités plus élevées chez l’adulte. Chez les patients adultes non traités ou traités tardivement, il a été rapporté une fréquence des anévrysmes coronaires de 19%, similaire à celle observée chez les enfants en l'absence de traitement.
TRAITEMENT
Le traitement de la MK à la phase aiguë vise à diminuer l’inflammation systémique et des artères coronaires pour limiter la formation d’anévrysmes et limiter le risque de complications à long-terme.
Stratégie thérapeutique conventionnelle
Le traitement à la phase aiguë de la maladie repose, selon les recommandations de l’AHA, sur une perfusion unique d’IgIV à la dose de 2 grammes par kilogramme, de préférence dans les 10 premiers jours de la fièvre. Il est recommandé d’associer à ce traitement de l’aspirine à la dose anti-inflammatoire de 80-100 mg/kg/j en 4 prises, jusqu’à 48-72 heures après obtention de l’apyrexie. L’aspirine pourrait en effet avoir un effet bénéfique à la phase aiguë de la maladie en raison de son action anti-inflammatoire mais son utilisation ne semble toutefois pas modifier l’incidence des anévrysmes coronaires. Une méta-analyse comparant des posologies d’aspirine de 80-120 mg/kg/j à des posologies de 30-50 mg/kg/j n’a pas mis en évidence de différence significative d’incidence des anévrysmes coronaires et certains recommandent de privilégier les posologies de 30-50 mg/kg/j, en raison d’une meilleure tolérance digestive, et ce jusqu’à obtention de l’apyrexie et diminution de l’inflammation. L’aspirine est ensuite diminuée à une dose anti-agrégante de 3 à 5 mg/kg/j en une prise pendant 6 à 8 semaines, qui sera poursuivie au long cours en cas d’anomalies coronaires persistantes. Une étude japonaise a comparé 71 patients traités par IgIV et aspirine (30 mg/kg/j) administrée de façon concomitante, et 111 patients chez qui l’aspirine a été débutée 24h après les IgIV. L’incidence des lésions coronaires était significativement différente entre les deux groupes, aucun anévrysme coronaire n’étant observé au-delà de 30 jours dans le 2ème groupe contre 6% dans le 1er. L’aspirine pourrait ainsi avoir un impact négatif initial sur la prévention du risque d’anévrysme coronaire par les IgIV (par une perturbation des fonctions immunologiques des lymphocytes T impliqués dans le mécanisme d’action des IgIV) et certaines équipes n’introduisent donc l’aspirine que 24h après le début des IgIV.
L’introduction des IgIV dans les 10 premiers jours de la fièvre permet de diminuer l’incidence des anévrysmes de 20-25 à 3-5%. Cependant, la proportion de patients présentant une résistance aux IgIV peut atteindre 20-25% et ces patients ont un risque accru de développer des anévrysmes coronaires en l’absence de traitement de 2ème ligne.
Stratégies évaluées chez les patients non-répondeurs aux IgIV
Une étude japonaise a évalué l’intérêt des bolus de méthylprednisolone chez 411 patients avec une fièvre persistante ou récurrente, 36h après une cure d’IgIV. Parmi les 63 patients non-répondeurs, 44 patients ont reçu des bolus de méthylprednislone sur 3 jours successifs et 19 ont reçu une cure supplémentaire d’IgIV. Une apyrexie rapide était obtenue chez 77% des patients recevant les bolus de méthylprednisolone et 63% des IgIV mais l’incidence des anévrysmes coronaires n’était pas significativement différente entre les 2 groupes (11% vs 10%). Plus récemment, une autre étude japonaise a comparé de manière rétrospective, chez 359 patients non-répondeurs à une première cure d’IgIV (2 g/kg), une nouvelle cure d’IgIV (1 ou 2 g/kg), un traitement par prednisolone (2 mg/kg/j jusqu’à normalisation de la CRP puis décroissance sur 15 jours), ou l’association des deux. Des anomalies des artères coronaires 1 mois après le traitement étaient significativement moins fréquemment retrouvées dans le groupe IgIV plus prednisolone (16%) comparativement au groupe IgIV (29%) et au groupe prednisolone (31%). De plus, le groupe traitement combiné avait moins de risque de non-réponse (12% vs 37%) que le groupe IgIV seules. Ces données suggèrent que l’association IgIV et prednisolone pourrait être supérieure aux IgIV seules ou à la prednisolone seule chez les patients non-répondeurs à une 1ère cure d’IgIV.
L’intérêt des anti-TNF-achez les patients non-répondeurs à une 1ère cure d’IgIV a également été évalué dans des études de faibles effectifs, en comparant l’efficacité de l’infliximab à celle d’une nouvelle cure d’IgIV, avec des résultats contrastés. Une première étude retrouvait une équivalence des deux stratégies, tandis qu’une autre retrouvait une régression plus rapide de la fièvre et une durée plus courte d’hospitalisation mais sans différence en terme d’événements cardiaques. Très récemment, une étude japonaise a évalué prospectivement l’intérêt de l’infliximab chez les patients non-répondeurs à une 1ère cure d’IgIV. Quarante-trois enfants ont été randomisés pour recevoir une nouvelle cure d’IgIV de 2 g/kg (n=32) ou de l’infiximab à la dose de 5 mg/kg (n=11). Le taux de réponse observé après une nouvelle cure d’IgIV était inférieur (mais sans atteindre la significativité statistique) à celui observé après infliximab (66% vs 90% ; P=0,09). Le groupe infliximab avait une durée de fièvre et d’hospitalisation plus courte, avec un profil de sécurité satisfaisant. Il n’y avait par contre pas de différence statistiquement significative entre les groupes IgIV et infliximab pour les anomalies coronaires (12% vs 9% ; P=0,4).
SURVEILLANCE
Surveillance et prise en charge après la phase aiguë
Le pronostic de la MK est largement conditionné par l’existence de complications cardiaques. Chez les patients sans complication initiale (régression de la fièvre sous traitement, absence d’anomalie cardiaque), une surveillance de l’échographie cardiaque transthoracique est recommandée par l’AHA à 2 semaines et à 6-8 semaines après le diagnostic de la maladie.
Surveillance et prise en charge à long-terme
Les recommandations de l’AHA stratifient les patients selon différents sous-groupes de risque croissant, allant des patients sans anévrysme en phase aiguë aux patients avec anévrysmes et sténoses coronaires. La prise en charge est stratifiée selon ce niveau de risque, allant de la simple surveillance clinique tous les 5 ans sans traitement médical associé, à une évaluation bi-annuelle avec des tests non-invasifs ou invasifs et un traitement médical plus ou moins lourd (aspirine, AVK, béta-bloquants, inhibiteurs de l’enzyme de conversion…). En France, la surveillance clinique simple n’est pas jugée nécessaire chez les patients sans anomalie coronaire.
PNDS sur la maladie de Kawasaki





