



Polyangéite microscopique
Un cours enregistré est également disponible en cliquant ici pour la présentation générale, et ici sur les immunothérapies ciblées au cours de la PAM.
Xavier Puéchal
La polyangéite microscopique (PAM) se définit comme une vascularite nécrosante systémique non granulomateuse touchant préférentiellement les vaisseaux de petit calibre. Elle est habituellement associée à des anticorps anti-cytoplasme des polynucléaires neutrophiles (ANCA), de fluorescence périnucléaire, dirigés contre la myéloperoxidase (MPO). Une glomérulonéphrite rapidement progressive et une hémorragie alvéolaire sont fréquentes.
CLASSIFICATION
La classification de l’American College of Rheumatology de la périartérite noueuse (PAN) de 1990 ne permettait pas de différencier la PAM de la PAN. La nomenclature de Chapel Hill a eu le mérite d’individualiser la PAM (1,2). Cette vascularite affecte préférentiellement les vaisseaux de petit calibre, i.e. les artérioles, capillaires et veinules (Figures 1 et 4) (2). Une glomérulonéphrite nécrosante pauci-immune et une capillarite pulmonaire s’observent au cours de la PAM tandis que dans la PAN, l’atteinte rénale est d’origine vasculaire et le poumon est épargné (Tableau 1). De plus, des ANCA dirigés contre la myéloperoxydase (MPO) sont présents chez la grande majorité des patients atteints de PAM et représentent un critère d’exclusion de la PAN. Les critères de classification de l’American College of Rheumatology / European League Against Rheumatism représentent maintenant les critères de référence (Tableau 2) (3). Ces critères ne doivent pas être utilisés à visée diagnostique.
EPIDEMIOLOGIE
La PAM est une maladie rare, touchant des sujets de toutes ethnies. Elle est ubiquitaire, avec un gradient Nord-Sud en Europe, la PAM étant plus fréquemment observée au Sud qu’au Nord. Elle survient volontiers chez des patients plus âgés que ceux atteints des autres vascularites nécrosantes systémiques. Son incidence en Europe varie de 2,6 à 10,2 par million d’habitants (4). Sa prévalence est estimée de 0 à 66 cas par million d’habitants en Europe dont 25 cas par million d’habitants en France et 86 cas par million au Japon (5,6).
PATHOGENIE
Des ANCA, dirigés contre les granules azurophiles des polynucléaires neutrophiles sont retrouvés dans 90 à 95 % des cas de PAM, principalement de fluorescence périnucléaire et dirigés contre la myéloperoxidase en ELISA (pANCA-MPO) (4). Le rôle des ANCA dans la pathogénie de la PAM semble plus important que dans la granulomatose avec polyangéite (GPA), au cours de laquelle le rôle de la protéinase 3 semble au-devant de la scène. Des souris invalidées pour le gène de la MPO auxquelles on transfecte des splénocytes de souris immunisées contre la MPO, développent une glomérulonéphrite extracapillaire et une hémorragie alvéolaire. Si l’on transfecte uniquement des IgG anti-MPO, les souris développent également des lésions proches de celles de la PAM (7). Chez l’homme, le rôle direct pathogène de ces ANCA est étayé in vivo par une observation de syndrome pneumo-rénal chez un nouveau-né après passage transplacentaire d’ANCA anti-MPO maternels (8). Des travaux expérimentaux ont montré la nécessité d’une activation de la voie alterne du complément pour que les lésions se développent. Les souris déficientes en facteur B ou en C5 ne développent pas de vascularite. L’efficacité clinique des anti-C5a a confirmé l’importance de cette activation. Après activation par des cytokines comme le TNF, la MPO des polynucléaires neutrophiles peut s’exprimer à leur surface et devenir accessible aux ANCA-MPO. Les ANCA favorisent alors leur adhésion à la surface des vaisseaux, provoquent la production et la libération de radicaux libres et leur dégranulation. Il en résulte un cascade inflammatoire aboutissant aux lésions inflammatoires vasculaires de la paroi des vaisseaux. D’autres mécanismes interviennent probablement comme un déséquilibre cytokinique et/ou d’expression de certaines molécules d’adhérence. Des anticorps anti-cellules endothéliales sont également détectables dans le sérum de patients atteints de PAM, avec un titre corrélé à l’activité de la maladie mais leur rôle reste incertain. Une prédisposition génétique est confortée par l’association avec HLA-DQ, qui est plus forte avec la spécificité anti-MPO des ANCA qu’avec le phénotype clinique de vascularite (PAM) (9).
MANIFESTATIONS CLINIQUES
Le début peut être très insidieux. Des arthralgies ou des hémoptysies peuvent survenir plusieurs mois ou années avant le diagnostic. Les principales manifestations cliniques au diagnostic de la PAM sont présentées dans le Tableau 3 avec leur fréquence respective (10-15). L’âge médian des patients est de 63,7 ans avec un sexe ratio équilibré (15).
Signes généraux
Une altération de l’état général est fréquente. Un amaigrissement et une fièvre se rencontrent chez la moitié des patients. Ces manifestations sont précoces et peuvent être inaugurales. Les signes généraux peuvent être isolés et l’apparition des autres manifestations conduit alors au diagnostic. A ce stade, la biopsie musculaire peut permettre le diagnostic.
Arthralgies
L’atteinte articulaire peut être inaugurale et touche presque la moitié des patients. Les arthralgies prédominent sur les grosses articulations. Les arthrites sont moins fréquentes. Elles sont non érosives.
Myalgies
Les myalgies sont présentes dans la moitié des cas. Elles sont intenses, diffuses, spontanées ou déclenchées par la pression. Parfois leur intensité peut en imposer pour une polymyosite mais les enzymes musculaires sont normales. Les patients sont parfois confinés au lit du fait de l’intensité des douleurs et de la fonte musculaire.
Atteinte rénale
L’atteinte rénale est une des caractéristiques majeures de la PAM sous forme d’une glomérulonéphrite extracapillaire pauci-immune rapidement progressive. Initialement insidieuse, elle survient chez les trois quarts des patients. Une hématurie microscopique et une protéinurie glomérulaire précèdent l’altération de la fonction rénale. Elles doivent être recherchées systématiquement. Plusieurs poussées frustres de PAM peuvent survenir avant la constatation d’une atteinte rénale majeure. En effet, les biopsies rénales objectivent souvent, à côté de lésions glomérulaires aiguës, des lésions cicatricielles anciennes laissent préjuger d’une mauvaise récupération de la fonction rénale malgré le traitement.
Atteinte pulmonaire
Une hémorragie alvéolaire, par capillarite pulmonaire, survient dans 12 à 29 % des PAM et peut révéler la maladie. Elle ne s’extériorise pas toujours. Les hémoptysies peuvent être modérées ou massives et responsables d’une détresse respiratoire, d’une anémie, voire d’un état de choc. Lorsque l’hémorragie alvéolaire s’accompagne d’une glomérulonéphrite rapidement progressive, elle s’intègre dans un syndrome pneumo-rénal, dont la PAM et la GPA sont les premières causes avant la vascularite anti membrane basale glomérulaire (Goodpasture). Les rechutes ne sont pas exceptionnelles et peuvent être mortelles (16). Une pneumopathie interstitielle fibrosante peut aussi précéder de plusieurs années les premières manifestations vasculaires. Elle figure parmi les nouveaux critères de classification de l’ACR / EULAR (3). Son pronostic dépend de la forme clinique. En cas de pneumopathie interstitielle commune, il y a une surmortalité qui persiste malgré les immunosuppresseurs administrés comme traitement de la vascularite (17).
Manifestations neurologiques
Atteinte du système nerveux périphérique. Les atteintes du système nerveux périphérique sont les plus fréquentes des manifestations neurologiques des vascularites. Elles engagent le pronostic fonctionnel. Une mononeuropathie multiple se manifeste avec une atteinte distale asymétrique, qui s’étend souvent par poussées successives. Des paresthésies et des douleurs peuvent précéder l’apparition d’un déficit moteur. Devant une neuropathie périphérique, un début brutal et un œdème segmentaire distal constituent des arguments en faveur d'une vascularite. Les nerfs touchés sont habituellement le nerf fibulaire commun, tibial voire plus rarement radial, ulnaire et/ou médian. L'amyotrophie apparaît rapidement. Une polyneuropathie distale, symétrique, sensitive ou sensitivo-motrice est plus rare. Le degré de récupération est imprévisible. Chez certains patients, une mononeuropathie multiple déficitaire sévère régresse totalement, tandis que d’autres gardent de lourdes séquelles motrices ou des douleurs neuropathiques résiduelles invalidantes. L’électromyogramme confirme le mécanisme axonal de la neuropathie en montrant des potentiels d’action moteurs et sensitifs absents ou diminués, contrastant avec des vitesses de conduction nerveuse normales ou modérément allongées.
Atteinte du système nerveux central. L’atteinte du système nerveux central est rare mais engage le pronostic vital. L’IRM peut détecter des hypersignaux multiples traduisant une vascularite cérébrale parfois sans traduction clinique. La présentation clinique varie selon la localisation et l’extension de l’atteinte vasculaire. Une hémiplégie, une comitialité localisée ou généralisée, voire des troubles cognitifs peuvent survenir.
Manifestations cutanées
Un purpura vasculaire est fréquent. Il résulte de l’atteinte des petits vaisseaux dermiques. Le purpura prédomine aux membres inférieurs. Chez un petit nombre de patients, un purpura bulleux ou des lésions vésiculeuses peuvent laisser place à des lésions nécrotiques. Une urticaire et des lésions annulaires fugaces peuvent s’associer au purpura. Des ulcérations, des nécroses cutanées, des hémorragies sous-unguéales sont possibles. La biopsie montre une vascularite des vaisseaux cutanés de petit calibre, parfois nécrosante, mais souvent ne visualise que des aspects de vascularite leucocytoclasique.
Manifestations gastro-intestinales
L’atteinte intestinale se manifeste par des douleurs abdominales, une hémorragie ou une perforation ischémique, surtout de l’intestin grêle. Elle est alors de mauvais pronostic.
Manifestations cardiovasculaires
Les manifestations cardiovasculaires sont rares au cours de la polyangéite microscopique, de même que les atteintes cardiaques, péricardiques ou myocardiques.
EXAMENS COMPLEMENTAIRES
Examens biologiques.
Un syndrome inflammatoire biologique est présent chez la majorité des patients. Une anémie d’origine inflammatoire peut être présente, mais elle est aussi parfois liée à un saignement digestif ou alvéolaire, extériorisé ou non. En cas d’atteinte rénale, l’hématurie microscopique est quasi constante avec une protéinurie glomérulaire mais rarement néphrotique. Le degré de dégradation de la fonction rénale dépend considérablement du recrutement. Le taux moyen de la créatininémie était de 574 µmol/l dans une série rénale (11) mais de 217 µmol/l dans notre large série multidisciplinaire (15).
Des ANCA sont mis en évidence chez les patients atteints de PAM, de fluorescence périnucléaire et de spécificité anti-MPO, dans 90 à 95 % des cas (Figure 7). Les tests par immuno-essai, comme l’ELISA, sont devenus plus sensibles et plus spécifiques que les tests par immunofluorescence, qu’ils vont remplacer pour la détection des ANCA et la confirmation de leur cible antigénique (18). De très rares patients pourraient avoir des ANCA dirigés contre la PR3 mais ces formes restent discutées et le diagnostic alternatif de GPA doit être envisagé. Ainsi, dans notre série de 378 patients atteints de PAM, l’ELISA trouvait des ANCA-MPO dans 92 % des cas, des anti-PR3 dans 4 % des cas et l’absence d’ANCA dans 4 % des cas (15). Dans un contexte de vascularite systémique, la présence d’ANCA anti-MPO est un critère d’exclusion de la PAN et un argument en faveur d’une PAM, qui figure dans les critères de classification de l’ACR / EULAR (Tableaux 1 et 2) (3). Néanmoins, la valeur diagnostique des ANCA anti-MPO dans la PAM est moins bonne que celle des ANCA anti-PR3 au cours de la GPA. En effet, des ANCA peuvent être mis en évidence dans une grande variété de maladies inflammatoires, de cible antigénique variée, et ne sont spécifiques ni de vascularite, ni de glomérulonéphrite.
Histologie rénale
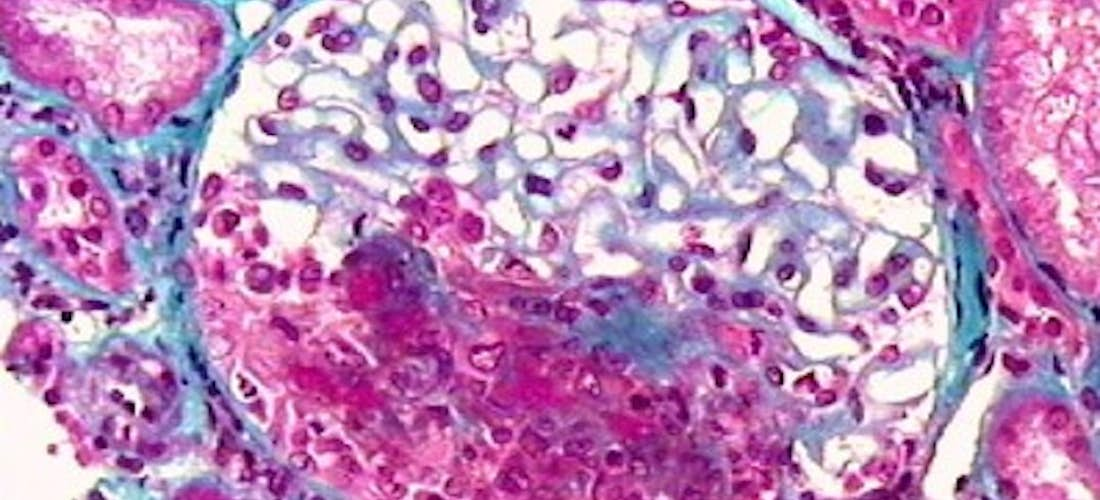
En cas d’atteinte rénale, l’histologie révèle une glomérulonéphrite nécrosante et thrombosante, segmentaire et focale. Une prolifération extracapillaire avec des croissants est quasi constante et touche souvent plus de 60 % des glomérules (Figure 5). L’immunofluorescence ne met pas en évidence de dépôts de complexes immuns. Les lésions histologiques permettent d’analyser les possibilités de récupération de la fonction rénale.
EVOLUTION ET PRONOSTIC
Rechutes
Une fois la rémission complète obtenue, les rechutes sont fréquentes au cours de la PAM et surviennent avec un délai médian de 2 ans après le diagnostic. Dans nos séries, des rechutes ont affecté entre un tiers et la moitié des patients (15,19,20). Les rechutes sont habituellement moins sévères que la poussée initiale et prises en charge rapidement chez un patient éduqué et alerté par la réapparition des signes de sa maladie. La survie sans rechute à 5 ans, au cours des études prospectives, était de 45 % et 53 % pour les formes de bon et mauvais pronostic selon le five factor score (FFS), respectivement (19,20). Le taux de rechute est amené à diminuer du fait de l’arrivée de traitement d’entretien plus efficaces comme le rituximab.
Décès
La survie globale des patients à 5 ans est d’environ 85 % et 94 % pour les patients diagnostiqués après 2010 (15). La plupart des décès survient chez des patients ayant un ou plusieurs facteurs de mauvais pronostic selon le FFS. Dans nos séries prospectives, le taux de survie globale à 5 ans était de 95 % et 65 % pour les formes de bon et mauvais pronostic, respectivement (19,20). Il existe une surmortalité dans les 18 premiers mois de la vascularite mais qui tend à disparaître ultérieurement. Dans la PAM, la principale cause de décès est l’insuffisance rénale et l’hémorragie alvéolaire, souvent en rapport avec une vascularite non contrôlée avec défaillance multiviscérale, favorisée par un âge élevé. Les infections représentent une autre cause importante de mortalité et sont favorisées par la corticothérapie et/ou les immunosuppresseurs. Les septicémies surviennent volontiers lors du traitement d’induction. Les infections virales sont plus tardives et reflètent l’immunodépression induite par la thérapeutique prescrite pour contrôler la maladie. Une infection par Pneumocystis jirovecii peut survenir au cours de la PAM et doit être prévenue par le cotrimoxazole.
Pronostic
Pour aider le clinicien à proposer la thérapeutique la plus appropriée et éviter notamment le sur-traitement des vascularites nécrosantes systémiques, nous avons proposé un score pronostique FFS (21), qui a été actualisé (22). Il tient compte des manifestations associées à une surmortalité, à savoir l’existence d’un âge supérieur à 65 ans, d'une cardiomyopathie spécifique, de manifestations gastro-intestinales, d’une insuffisance rénale définie par une créatininémie stabilisée supérieure à 150 µmol/l ainsi que de l’absence de manifestations ORL pour la GPA et la granulomatose éosinophilique avec polyangéite (Tableau 4). En l’absence de ces facteurs pronostiques défavorables, le FFS est nul et la mortalité est de 9 % à 5 ans (22). Lorsqu’un facteur est présent, le FFS est de 1 et la mortalité est de 21 % à 5 ans. Lorsque plus d’un facteur est présent, la mortalité est de 40 %.
Dans le PNDS concernant la GPA et la PAM, dans un souhait d’harmonisation de la prise en charge avec les pratiques internationales, l’utilisation de la notion de « maladie sévère » et de « maladie non sévère » a été préférée au FFS pour adapter la stratégie thérapeutique (23). Ainsi, les manifestations « sévères » regroupent (liste non restrictive à ces seules manifestations et à adapter à l’avis du clinicien) : cardiomyopathie spécifique, ischémie mésentérique, hémorragie intra-alvéolaire sévère avec détresse respiratoire, insuffisance rénale rapidement progressive, accident vasculaire cérébral spécifique, atteinte de(s) paire(s) crânienne(s), mononeuropathie multiple avec atteinte motrice sévère...
TRAITEMENT DE LA POLYANGEITE MICROSCOPIQUE
Une synthèse des recommandations thérapeutiques proposées par le PNDS en 2019 pour le traitement des vascularites associées aux ANCA se trouve Tableau 5 (23). La prise en charge thérapeutique comprend un traitement d’induction dont le but est d’obtenir une rémission complète, obtenue dans plus de 80 % des cas en 3 mois, suivi d’un traitement d’entretien dont le but est d’éviter une rechute de la vascularite. Le traitement est débuté souvent en urgence, en milieu spécialisé, de façon adaptée à la gravité de la maladie. La base de la thérapeutique reste la corticothérapie, qui est associée en première intention aux immunosuppresseurs uniquement dans les formes sévères, notamment définies par un FFS au moins égal à 1.
Traitement d’induction
Les corticoïdes
Les corticoïdes restent la base du traitement de la PAM. Les bolus de méthylprednisolone sont souvent administrés à la posologie de 15 mg/kg pendant 1 à 3 jours. Ils sont très largement utilisés, particulièrement en cas de menace immédiate du pronostic vital ou à la phase d'extension d'une polyneuropathie, en raison de leur efficacité rapide et relative innocuité. La corticothérapie orale est initialement administrée à la posologie de 1 mg/kg/jour de prednisone en une prise unique matinale. Des schémas de décroissance rapide ont été récemment validés (24-26) mais parfois au prix d’une majoration de la fréquence des rechutes. Les recommandations actuelles sont d’atteindre 20 mg/j à 6 semaines, 10 mg/j à 3 mois et 5 mg/j avant 5 mois, à maintenir jusqu’à 1 an [schéma PEXIVAS bras court (25)] (27-29).
Le cyclophosphamide
Le cyclophosphamide ne doit pas être prescrit systématiquement en première intention chez tous les patients atteints de PAM. La décision doit tenir compte des localisations viscérales, de leur sévérité et de l’activité de la maladie. Le cyclophosphamide ne se discute en première intention que dans le traitement d’induction de la PAM sévère. Bien que le cyclophosphamide par voie orale (2 mg/kg/jour) soit un traitement efficace au cours des vascularites, son rapport efficacité/toxicité est faible. Les effets indésirables d'une administration quotidienne de cyclophosphamide sont la possibilité de cystite hémorragique, d'hypoplasie médullaire, d'insuffisance ovarienne et/ou de néoplasie (cancer de la vessie et lymphome). Les infections représentent également une cause majeure de mortalité. Pour diminuer ces effets indésirables, les perfusions de cyclophosphamide se sont imposées dans le traitement des vascularites nécrosantes systémiques sévères et doivent être privilégiées à l'administration orale continue (30-32). Elles permettent d’administrer une dose cumulative inférieure exposant le patient à une toxicité moindre que lors de l’utilisation de la voie orale continue avec une efficacité comparable à 6 mois sur le taux de mise en rémission. Le cyclophosphamide est prescrit en perfusions de 0,6 g/m2. Après 3 perfusions à 14 jours d’intervalle, l’administration est poursuivie à 0,7 g/m2 toutes les trois semaines jusqu’à obtention d’une rémission complète. Habituellement, 6 bolus sont nécessaires en 3 mois avant le relais par un traitement d’entretien. La posologie est à adapter à l’âge et à la fonction rénale. Une hydratation abondante et l’utilisation de mesna (UromitexanÒ) doivent être associées aux bolus.
Le rituximab
Le rituximab ne se discute en première intention que dans la PAM sévère. Ce biomédicament a une efficacité comparable au cyclophosphamide, comme traitement d’induction des formes sévères de PAM, pour lesquels il représente une alternative. Dans un essai randomisé, le rituximab (anti-CD20) a montré qu’il n’était pas inférieur au cyclophosphamide pour induire une rémission avec une tolérance comparable, au cours de PAM sévères ou de GPA (33). Les patients traités par rituximab (4 perfusions hebdomadaires de 375 mg/m2) avaient un taux de mise en rémission complète non inférieur à celui de ceux traités par cyclophosphamide relayé par l’azathioprine. Le profil de tolérance était comparable à court terme (33-35). En cas de rechute, le rituximab était associé à un meilleur taux de mise en rémission (33). Lorsqu’une rémission n’était pas obtenue par le cyclophosphamide, le rituximab permettait alors souvent de l’obtenir. En cas de rechute ou d’échec du cyclophosphamide, le rituximab est donc privilégié. Un délai de 3 mois est parfois nécessaire pour obtenir un effet thérapeutique maximal. Le rituximab a ainsi obtenu une autorisation américaine et européenne de mise sur le marché comme traitement d’induction des PAM et GPA sévères, en association aux corticoïdes.
Autres immunosuppresseurs
Le mycophénolate mofétil, l’azathioprine, le méthotrexate et d’autres cytotoxiques ont été essayés dans la PAM.
Un essai de non infériorité a montré une certaine efficacité du mycophénolate mofétil dans le traitement d’induction de la PAM mais qui semble inférieure à celle du cyclophosphamide et expose notamment à davantage de rechutes (36). Ce traitement ne peut donc être recommandé en première intention. Une étude contrôlée réalisée en traitement d’entretien a aussi montré que le mycophénolate mofétil était moins efficace que l’azathioprine pour prévenir les rechutes (37).
Au cours de la PAM de bon pronostic (FFS=0), l’azathioprine en traitement d’induction en association à la corticothérapie n’augmente pas le contrôle de ces maladies (38). L’azathioprine et le méthotrexate ont aussi été largement utilisés comme traitement d’entretien de la PAM sévère avec des résultats comparables (39). Néanmoins, des rechutes restaient très fréquentes à l’arrêt du traitement ou à la diminution des doses.
L’utilisation d’autres immunosuppresseurs ne repose pas sur les résultats d’études prospectives et ne peut donc pas être recommandée.
Traitements adjuvants
En raison de l’immunosuppression iatrogène, une prophylaxie contre les infections opportunistes comme Pneumocystis jirovecii est recommandée (23,27). Elle repose habituellement sur le triméthoprime-sulfaméthoxazole (par exemple, BactrimÒ forte 800 mg : 1 comprimé, 3 fois par semaine ou BactrimÒ 400 mg : 1 comprimé/jour). Si l’incidence des pneumocystoses reste rare, des études rétrospectives (40,41) et surtout prospectives (33,42,43) ont montré que les patients recevant cette prophylaxie présentaient aussi un moindre taux global d’infection sévère.
Les antalgiques, la prévention des escarres et la rééducation s’imposent dans les mononeuropathies multiples. Les inhibiteurs du système rénine angiotensine (de l’enzyme de conversion de l’angiotensine ou des récepteurs de l’angiotensine II) sont prescrits à visée de protection néphronique en cas d’atteinte rénale. Pour les patients avec manifestations digestives, le traitement d’attaque est administré par voie veineuse pour éviter les problèmes inhérents à une mauvaise absorption. Une nutrition parentérale est mise en route en cas d’atteinte digestive sévère avec cachexie rapidement progressive. La correction d’un état nutritionnel général médiocre peut aider à diminuer l’incidence des infections induites par les traitements cytotoxiques. En cas d’angor ou d’insuffisance cardiaque, les investigations appropriées sont utiles pour apprécier le mécanisme lésionnel et détecter une étiologie surajoutée comme l’athérosclérose qui peut être traitée spécifiquement. L'ostéoporose cortisonique doit être combattue, selon les recommandations habituelles, par une activité physique régulière, une alimentation riche en calcium, une supplémentation vitamino-calcique et éventuellement un bisphosphonate. La rééducation d’une neuropathie périphérique et la confection d’attelle en cas de déficit important permettent souvent une reprise plus rapide de la mobilité. L’augmentation de la fréquence des complications cardio-vasculaires au cours des vascularites associées aux ANCA justifie un dépistage et une prise en charge des facteurs de risque cardio-vasculaire associés (44). Enfin, l’importance du retentissement psychologique des vascularites et de sa prise en charge ne doit pas être sous-estimée (45).
Traitement d’entretien
Le rituximab est devenu le traitement d’entretien de première intention des formes sévères de PAM.
Il a supplanté les immunosuppresseurs conventionnels d’entretien (46). En effet, comme traitement d’entretien des formes sévères de PAM ou GPA, après obtention de la rémission, le rituximab diminue très significativement le taux de rechute par rapport au traitement immunosuppresseur conventionnel avec un profil de tolérance acceptable (46,47). Le rituximab d’entretien est débuté dans les 4 semaines après le dernier bolus de cyclophosphamide ou 4 à 6 mois après le début d’un traitement d’induction par le rituximab. Il s’administre sous forme de perfusions de 500 mg tous les 6 mois. Il a été montré que la perfusion à J15, initialement proposée dans l’étude princeps (46), pouvait être omise (48). La durée du traitement d’entretien du rituximab varie de 2 ans (4 perfusions en 18 mois) à 4 ans (8 perfusions en 42 mois), surtout chez les patients à fort risque de rechute comme ceux ayant déjà rechuté ou ceux avec des ANCA-PR3 (49). Néanmoins, les études de suivi ont montré l’absence de bénéfice à long terme de prolonger à 4 ans le traitement d’entretien (50). Un autre schéma d’entretien est éventuellement possible en perfusant le rituximab uniquement en cas de réapparition des CD19 ou d’ascension du taux des ANCA vérifiés tous les 3 mois (51) mais son efficacité est moindre. Une concentration sanguine de rituximab, inférieure à 4 μg/ml, à 3 mois de l’initiation du traitement d’entretien, s’est révélée être un facteur de risque indépendant de rechute majeure ultérieure (52). De nouveaux protocoles d’administration devront évaluer si le dosage sanguin du rituximab permet effectivement d’améliorer la prise en charge. Ce biomédicament a obtenu fin 2018 une extension américaine et européenne d’autorisation de mise sur le marché comme traitement d’entretien de ces formes, dont il représente le traitement de référence.
Indications thérapeutiques dans la PAM sans facteur de mauvais pronostic
Pour les formes de bon pronostic, la corticothérapie isolée reste le traitement de première intention (23). La corticothérapie seule est efficace et peut suffire à contrôler la maladie sans rechute ultérieure à 5 ans dans presque la moitié des cas de PAM sans facteur pronostique défavorable (FFS=0) (19,53). La survie à 8 ans reste excellente (86 %) mais environ un patient sur deux nécessite l’adjonction ultérieure d’un immunosuppresseur (19). Chez les patients non répondeurs à la seule corticothérapie ou en rechute, l’addition secondaire du cyclophosphamide ou de l’azathioprine permet habituellement de contrôler la situation. La durée totale recommandée de la corticothérapie est de 18 à 24 mois (23).
Indications thérapeutiques dans la PAM avec facteur de mauvais pronostic
Le traitement d’induction associe une forte corticothérapie en association aux perfusions de cyclophosphamide ou de rituximab. Après l’obtention d’une rémission, un traitement d’entretien est nécessaire pour éviter les rechutes. Le rituximab comme traitement d’entretien a relayé au second plan les traitements immunosuppresseurs conventionnels (azathioprine ou méthotrexate) (46).
Place de la surveillance du taux des ANCA
Une majoration du taux des ANCA en rémission se voit plus souvent chez les patients qui vont rechuter mais elle n’est pas suffisamment informative pour le prévoir à titre individuel (54). Une intensification thérapeutique n’est pas non plus dénuée de risque. Il est ainsi recommandé de ne pas envisager de majoration thérapeutique en raison d’une élévation ou d’une persistance isolée des ANCA mais de majorer la surveillance (23,27).
Place des perfusions de gammaglobulines
L’intérêt des IgIV a été suscité par leur efficacité dans la maladie de Kawasaki où elles préviennent le développement des anévrysmes des artères coronaires. Les IgIV ont été essentiellement utilisées dans la GPA et la PAM en rechute. La dose totale de 2 g/kg est administrée en 2 perfusions (1g/kg/j ; J1 et J2) ou repartie sur 5 jours, surtout en cas d'atteinte rénale (0,4 g/kg/j ; J1 à J5). Les résultats d’études ouvertes et d’une étude contrôlée sont encourageants (55,56). Même si l’efficacité est inconstante et parfois transitoire, elles peuvent néanmoins permettre de passer un cap comme traitement adjuvant, en association avec une immunosuppression, chez les patients atteints de vascularite n'ayant pas répondu au traitement de première ligne de référence (vascularite réfractaire) et/ou surtout en cas d’infection surajoutée. Leur prescription doit être discutée avec un centre de compétences ou de référence, afin de pouvoir ensuite en évaluer la prescription.
Place des échanges plasmatiques
Des échanges plasmatiques étaient souvent réalisés dans les formes sévères de vascularites associées aux ANCA avec insuffisance rénale rapidement progressive pour diminuer le taux de patients en insuffisance rénale terminale. Dans une étude européenne randomisée contrôlée, ils se sont montrés significativement supérieurs aux bolus de méthylprednisolone chez des patients avec une créatininémie initiale supérieure à 500 mmol/l pour diminuer le nombre de patients dialysés à 3 et 12 mois (57). Néanmoins, ils n’affectaient pas la survie globale et le bénéfice rénal ne se maintenait pas sur le long terme. L’étude PEXIVAS, essai randomisé évaluant en première intention l’intérêt des échanges plasmatiques en cas d’atteinte rénale ou d’hémorragie alvéolaire chez plus de 700 patients atteints de vascularite associée aux ANCA, n’a pas montré de bénéfice des échanges plasmatiques sur un critère composite associant décès ou évolution vers l’insuffisance rénale définitive (25). Depuis les résultats de cette étude, on a tendance à ne proposer les échanges que dans les formes sévères réfractaires à un traitement médical bien conduit (en seconde intention) ou en cas d’hémorragie alvéolaire massive (23).
Place des inhibiteurs du C5a
Les inhibiteurs du C5a sont prometteurs et pourraient permettre de diminuer voire de se passer de la corticothérapie. L’avacopan est un premier antagoniste du récepteur du C5A qui s’administre par voie orale. Une étude de phase III, en association au cyclophosphamide ou au rituximab, a démontré sa non infériorité à 6 mois et sa supériorité à 12 mois, comme traitement d’induction de la granulomatose avec polyangéite ou de la PAM, par rapport à une corticothérapie brève de 6 mois pour le taux de mise en rémission complète (58). L’avacopan pourrait avoir un bénéfice supplémentaire sur la fonction rénale. Ce médicament a obtenu une autorisation de mise sur le marché et son remboursement. D’autres inhibiteurs du C5 et d’autres modulateurs de la voie alterne du complément sont en cours d’investigation. Leur place au sein de l’arsenal thérapeutique reste à définir.
Autres traitements
Dans certaines formes particulièrement réfractaires de vascularite associée aux ANCA, d’autres options thérapeutiques existent. Des essais en cours évaluent des traitements ciblant la réponse immune adaptative aberrante (traitements dirigés contre les lymphocytes B ou T) et ceux dirigés contre des éléments de la réponse innée comme le complément, les monocytes, ou les neutrophiles (59).
Prise en charge d’une hémorragie alvéolaire
Une hémorragie alvéolaire est habituellement traitée par l’association de corticoïdes et de cyclophosphamide ou de rituximab, voire d’échanges plasmatiques si elle est très importante ou en l’absence d’amélioration avec un traitement médical bien conduit (23).
Le tableau clinique des formes fulminantes de PAM est souvent un syndrome pneumo-rénal. Le traitement d’une hémorragie alvéolaire massive nécessite en urgence un remplissage volémique et une assistance hémodynamique et respiratoire. La détérioration de la fonction rénale nécessite souvent l’épuration extrarénale. Les échanges plasmatiques ont un rôle dans ces formes avec atteinte sévère, surtout si elles ne répondent pas très rapidement à un traitement bien conduit. Néanmoins, le pronostic des formes suraiguës de PAM est sombre. Deux tiers des décès sont imputables à une vascularite active compliquée d’insuffisance rénale et d’hémorragie alvéolaire ou aux effets indésirables des thérapeutiques. Dans les cas difficiles, il est conseillé de discuter chaque observation avec les centres de référence et/ou de compétences. Ils proposent aussi la mise à disposition d’informations actualisées sur les critères diagnostiques, les outils de suivi, les recommandations thérapeutiques et les protocoles en cours (www.vascularites.org).
A RETENIR
Quand évoquer le diagnostic de polyangéite microscopique ?
Le diagnostic doit être évoqué devant les signes suivants d’autant plus qu’ils sont associés :
- Altération de l’état général
- Neuropathie périphérique
- Arthralgies d’allure inflammatoire
- Myalgies
- Purpura vasculaire
- Atteinte rénale glomérulaire
Comment confirmer le diagnostic de polyangéite microscopique ?
Diagnostic positif : l’histologie confirme le diagnostic de vascularite à partir d’un prélèvement cutané, musculaire, neuromusculaire, voire rénal. Le choix du prélèvement est orienté par le tableau clinique.
Diagnostic d’extension : toutes les manifestations classiques de la maladie sont recherchées (atteintes cutanées, neurologiques, rénales, cardiaques…) par l’interrogatoire, l’examen clinique et les investigations appropriées.
Diagnostic de gravité : Le score pronostique FFS (Five Factor Score) conditionne le pronostic et les indications thérapeutiques (Tableau 4). Les manifestations « sévères » regroupent cardiomyopathie spécifique, ischémie mésentérique, hémorragie intra-alvéolaire sévère avec détresse respiratoire, insuffisance rénale rapidement progressive, accident vasculaire cérébral spécifique, atteinte de(s) paire(s) crânienne(s), mononeuropathie multiple avec atteinte motrice sévère...
Diagnostic différentiel entre polyangéite microscopique et périartérite noueuse : La présence d’une glomérulonéphrite, d’une hémorragie alvéolaire ou d’ANCA oriente vers une PAM.
Références
1 Jennette JC, Falk RJ, Andrassy K, et al: Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum 1994;37:187-92.
2 Jennette JC, Falk RJ, Bacon PA, et al: 2012 revised international Chapel Hill consensus conference nomenclature of Vasculitides. Arthritis Rheum 2013;65:1-11.
3 Suppiah R, Robson JC, Grayson PC, et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology classification criteria for microscopic polyangiitis. Ann Rheum Dis 2022;81:321-6.
4 Kitching AR, Anders HJ, Basu N, et al: ANCA-associated vasculitis. Nat Rev Dis Primers 2020 27;6:71.
5 Watts RA, Lane S, Scott DG: What is known about the epidemiology of the vasculitides? Best Pract Res Clin Rheumatol 2005;19:191-207.
6 Mahr A, Guillevin L, Poissonnet M, Aymé S: Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener's granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: a capture-recapture estimate. Arthritis Rheum 2004;51:92-9.
7 Xiao H, Heeringa P, Hu P, et al: Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest 2002;110:955-63.
8 Bansal PJ, Tobin MC: Neonatal microscopic polyangiitis secondary to transfer of maternal myeloperoxidase-antineutrophil cytoplasmic antibody resulting in neonatal pulmonary hemorrhage and renal involvement. Ann Allergy Asthma Immunol 2004;93:398-401.
9 Lyons PA, Rayner TF, Trivedi S, et al: Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med 2012;367:214-23.
10 Serra A, Cameron JS, Turner DR, et al: Vasculitis affecting the kidney: presentation, histopathology and long-term outcome. Q J Med. 1984;53:181-207.
11 Savage CO, Winearls CG, Evans DJ, Rees AJ, Lockwood CM. Microscopic polyarteritis: presentation, pathology and prognosis. Q J Med 1985;56:467-83.
12 D'Agati V, Chander P, Nash M, Mancilla-Jimenez R: Idiopathic microscopic polyarteritis nodosa: ultrastructural observations on the renal vascular and glomerular lesions. Am J Kidney Dis 1986;7:95-110.
13 Adu D, Howie AJ, Scott DG, Bacon PA, McGonigle RJ, Micheal J: Polyarteritis and the kidney. Q J Med 1987;62:221-37.
14 Guillevin L, Durand-Gasselin B, Cevallos R: Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum 1999;42:421-30.
15 Nguyen Y, Pagnoux C, Karras A, et al: Microscopic polyangiitis: Clinical characteristics and long-term outcomes of 378 patients from the French Vasculitis Study Group Registry. J Autoimmun 2020;112:102467.
16 Lauque D, Cadranel J, Lazor R, et al: Microscopic polyangiitis with alveolar hemorrhage. A study of 29 cases and review of the literature. Groupe d'Etudes et de Recherche sur les Maladies "Orphelines" Pulmonaires (GERM"O"P). Medicine (Baltimore) 2000;79:222-33.
17 Maillet T, Goletto T, Beltramo G, et al: Usual interstitial pneumonia in ANCA-associated vasculitis: A poor prognostic factor. J Autoimmun 2020;106:102338.
18 Bossuyt X, Cohen Tervaert JW, et al: Revised 2017 international consensus on testing of ANCAs in granulomatosis with polyangiitis and microscopic polyangiitis. Nat Rev Rheumatol 2017;13:683-692.
19 Samson M, Puéchal X, Devilliers H, et al: Long-term follow-up of a randomized trial on 118 patients with polyarteritis nodosa or microscopic polyangiitis without poor-prognosis factors. Autoimmun Rev 2014;13:197-205.
20 Samson M, Puéchal X, Mouthon L, et al: Microscopic polyangiitis and non-HBV polyarteritis nodosa with poor-prognosis factors: 10-year results of the prospective CHUSPAN trial. Clin Exp Rheumatol 2017;35 Suppl 103:176-184.
21 Guillevin L, Lhote F, Gayraud M, et al: Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore) 1996;75:17-28.
22 Guillevin L, Pagnoux C, Seror R, Mahr A, Mouthon L, Toumelin PL; French Vasculitis Study Group (FVSG): The Five-Factor Score revisited: assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore) 2011;90:19-27.
23 Protocole National de Diagnostic et de Soins: Vascularites nécrosantes systémiques (périartérite noueuse et vascularites associées aux ANCA). https://www.has-sante.fr/upload/docs/application/pdf/2019-06/pnds_vns.pdf
24 Miloslavsky EM, Niles JL, Wallace ZS, et al: Reducing glucocorticoid duration in ANCA-associated vasculitis: A pilot trial. Semin Arthritis Rheum 2018;48:288-92.
25 Walsh M, Merkel PA, Peh CA, et al: Plasma exchange and glucocorticoids in severe ANCA-associated vasculitis. N Engl J Med 2020;382:622-631.
26 Furuta S, Nakagomi D, Kobayashi Y, et al: Effect of reduced-dose vs high-dose glucocorticoids added to rituximab on remission induction in ANCA-associated vasculitis: A randomized clinical trial. JAMA 2021;325:2178-87.
27 Chung SA, Langford CA, Maz M, et al: 2021 American College of Rheumatology/Vasculitis Foundation guideline for the management of antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheumatol 2021;73:1366-83.
28 Hellmich B, Sanchez-Alamo B, Schirmer JH, et al: EULAR recommendations for the management of ANCA-associated vasculitis: 2022 update. Ann Rheum Dis 2024;83:30-47.
29 Kidney Disease, Improving Global Outcomes (KDIGO) ANCA Vasculitis Work Group: KDIGO 2024 clinical practice guideline for the management of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Kidney Int 2024;105:S71-S116.
30 Guillevin L, Cordier JF, Lhote F, et al: A prospective, multicenter, randomized trial comparing steroids and pulse cyclophosphamide versus steroids and oral cyclophosphamide in the treatment of generalized Wegener's granulomatosis. Arthritis Rheum 1997;40:2187-98.
31 Haubitz M, Schellong S, Göbel U, et al: Intravenous pulse administration of cyclophosphamide versus daily oral treatment in patients with antineutrophil cytoplasmic antibody-associated vasculitis and renal involvement: a prospective, randomized study. Arthritis Rheum 1998;41:1835-44.
32 de Groot K, Harper L, Jayne DR, et al: Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med 2009;150:670-80.
33 Stone JH, Merkel PA, Spiera R, et al: Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010; 363: 221-32.
34 Jones RB, Cohen Tervaert JW, et al: Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med 2010; 363: 211-20.
35 Specks U, Merkel PA, Seo P, et al: Efficacy of remission-induction regimens for ANCA-associated vasculitis. N Engl J Med 2013; 369: 417-27.
36 Jones RB, Hiemstra TF, Ballarin J, et al: Mycophenolate mofetil versus cyclophosphamide for remission induction in ANCA-associated vasculitis: a randomised, non-inferiority trial. Ann Rheum Dis 2019;78:399‑405.
37 Hiemstra TF, Walsh M, Mahr A, et al: Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled trial. JAMA 2010;304:2381-8.
38 Puéchal X, Pagnoux C, Baron G, et al: Adding azathioprine to remission-induction glucocorticoids for eosinophilic granulomatosis with polyangiitis (Churg-Strauss), microscopic polyangiitis, or polyarteritis nodosa without poor prognosis factors: A randomized, controlled trial. Arthritis Rheumatol 2017;69:2175-86.
39 Pagnoux C, Mahr A, Hamidou MA, et al: Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med 2008;359:2790-803.
40 Kronbichler A, Kerschbaum J, Gopaluni S, et al: Trimethoprim-sulfamethoxazole prophylaxis prevents severe/life-threatening infections following rituximab in antineutrophil cytoplasm antibody-associated vasculitis. Ann Rheum Dis 2018;77:1440-7.
41 Mendel A, Behlouli H, Vinet É, Curtis JR, Bernatsky S. Trimethoprim sulfamethoxazole prophylaxis and serious infections in granulomatosis with polyangiitis treated with rituximab. Rheumatology (Oxford) 2025;64:2041-9.
42 Stegeman CA, Tervaert JW, de Jong PE, Kallenberg CG. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener's granulomatosis. Dutch Co-Trimoxazole Wegener Study Group. N Engl J Med 1996;335:16-20.
43 Zycinska K, Wardyn KA, Zielonka TM, Krupa R, Lukas W. Co-trimoxazole and prevention of relapses of PR3-ANCA positive vasculitis with pulmonary involvement. Eur J Med Res 2009;14 Suppl 4:265-7.
44 Morgan MD, Turnbull J, Selamet U, et al: Increased incidence of cardio-vascular events in patients with antineutrophil cytoplasmic antibody-associated vasculitides. Arthritis Rheum 2009;60:3493-500.
45 Koutantji M, Pearce S, Harrold E. Psychological aspects of vasculitis. Rheumatology (Oxford) 2000;39:1173-9.
46 Guillevin L, Pagnoux C, Karras A, et al: Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med 2014;371:1771-80.
47 Smith R, Jayne D, Merkel PA, et al: Extended follow-up of patients recruited to a randomized, controlled trial of rituximab versus azathioprine after induction of remission with rituximab for patients with ANCA-associated vasculitis and relapsing disease [abstract]. Arthritis Rheumatol 2020; 72:Supplement10:2052.
48 Charles P, Dechartres A, Terrier B, et al. Reducing the initial number of rituximab maintenance-therapy infusions for ANCA-associated vasculitides: randomized-trial post-hoc analysis. Rheumatology (Oxford) 2020;59:2970-75.
49 Charles P, Perrodeau É, Samson M, et al: Long-term rituximab use to maintain remission of antineutrophil cytoplasmic antibody-associated vasculitis: A randomized trial. Ann Intern Med 2020;173:179-87.
50 Delestre F, Charles P, Karras A, et al. Rituximab as maintenance therapy for ANCA-associated vasculitides: pooled analysis and long-term outcome of 277 patients included in the MAINRITSAN trials. Ann Rheum Dis 2024;83:233-41.
51 Charles P, Terrier B, Perrodeau É, et al: Comparison of individually tailored versus fixed-schedule rituximab regimen to maintain ANCA-associated vasculitis remission: results of a multicentre, randomised controlled, phase III trial (MAINRITSAN2). Ann Rheum Dis 2018;77:1143-9.
52 Khoudour N, Delestre F, Jabot-Hanin F, et al. Association between plasma rituximab concentration and the risk of major relapse in antineutrophil cytoplasmic antibody-associated vasculitides during rituximab maintenance therapy. Arthritis Rheumatol 2023, doi: 10.1002/art.42556. Online ahead of print.
53 Ribi C, Cohen P, Pagnoux C, et al: Treatment of polyarteritis nodosa and microscopic polyangiitis without poor-prognosis factors: A prospective randomized study of one hundred twenty-four patients. Arthritis Rheum 2010;62:1186-97.
54 Tomasson G, Grayson PC, Mahr AD, LaValley M, Merkel PA. Value of ANCA measurements during remission to predict a relapse of ANCA-associated vasculitis: a meta-analysis. Rheumatology (Oxford) 2012;51:100–9.
55 Martinez V, Cohen P, Pagnoux C, et al: Intravenous immunoglobulins for relapses of systemic vasculitides associated with antineutrophil cytoplasmic autoantibodies: results of a multicenter, prospective, open-label study of twenty-two patients. Arthritis Rheum 2008;58:308-17.
56 Jayne DR, Chapel H, Adu D, et al: Intravenous immunoglobulin for ANCA-associated systemic vasculitis with persistent disease activity. QJM 2000;93:433-9.
57 Jayne DR, Gaskin G, Rasmussen N, et al: Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. 2007; 18: 2180-8.
58 Jayne DRW, Merkel PA, Schall TJ, Bekker P; ADVOCATE Study Group: Avacopan for the treatment of ANCA-associated vasculitis. N Engl J Med 2021;384:599-609.
59 Prendecki M, McAdoo SP: New therapeutic targets in antineutrophil cytoplasm antibody-associated vasculitis. Arthritis Rheumatol 2021;73:361-70.
Présentation sur la granulomatose avec polyangéite et polyangéite microscopique

PNDS vascularites nécrosantes
Figure 1. Nomenclature révisée de la Conférence de Chapel Hill
Figure 7. ANCA IF
Figures 4. Nomenclature révisée de la Conférence de Chapel Hill
Figures 5. HISTOLOGIE VASCULARITES
Table 1
Table 2
Table 3
Table 4
Table 5




